Article Text

Abstract
Complex I deficiency is the most frequent mitochondrial disorder presenting in childhood, accounting for up to 30% of cases. As with many mitochondrial disorders, complex I deficiency is characterised by marked clinical and genetic heterogeneity, leading to considerable diagnostic challenges for the clinician, not least because of the involvement of two genomes. The most prevalent clinical presentations include Leigh syndrome, leukoencephalopathy and other early-onset neurodegenerative disorders; fatal infantile lactic acidosis; hypertrophic cardiomyopathy; and exercise intolerance. Causative genetic defects may involve the seven mitochondrial-encoded or 38 nuclear-encoded subunits of the enzyme, or any of an increasing number of assembly factors implicated in the correct biosynthesis of complex I within the inner mitochondrial membrane. In this review, we discuss recent advances in knowledge of the structure, function and assembly of complex I and how these advances, together with new high-throughput genetic screening techniques, have translated into improved genetic diagnosis for affected patients and their families. Approximately 25% of cases have mitochondrial DNA mutations, while a further ∼25% have mutations in a nuclear subunit or in one of nine known assembly factors. We also present a systematic review of all published cases of nuclear-encoded complex I deficiency, including 117 cases with nuclear subunit mutations and 55 with assembly factor mutations, and highlight clinical, radiological and biochemical clues that may expedite genetic diagnosis.
Statistics from Altmetric.com
Introduction
Complex I (nicotinamide adenine dinucleotide (NADH):ubiquinone oxidoreductase, Enzyme Commission number EC 1.6.5.3) is the first and largest enzyme of the mitochondrial respiratory chain (RC) and oxidative phosphorylation (OXPHOS) system, and plays critical roles in transferring electrons from reduced NADH to coenzyme Q10 (CoQ10, ubiquinone) and in pumping protons to maintain the electrochemical gradient across the inner mitochondrial membrane. This electrochemical gradient, generated by complexes I, III and IV, is subsequently harnessed by complex V (ATP synthase) to synthesise ATP from ADP and inorganic phosphate. Complex I is also the major site for the generation of reactive oxygen species (ROS), which are increasingly recognised to be important signalling molecules determining the health and fate of the mitochondrion and of the whole cell.
Isolated deficiency of complex I is the most commonly identified biochemical defect in childhood-onset mitochondrial disease, accounting for approximately a third of all cases of OXPHOS disorders.1 Complex I deficiency is clinically heterogeneous but the majority of affected individuals develop symptoms during the first year of life and have a rapidly progressive disease course, resulting in a fatal outcome in childhood. However, clinical presentations may vary, ranging from fatal neonatal lactic acidosis to infantile-onset Leigh syndrome, childhood-onset mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS) syndrome and, in some cases, adult-onset encephalomyopathic syndromes of variable severity. Presentation with single organ involvement is also recognised, for example, isolated hypertrophic cardiomyopathy (HCM) or Leber's hereditary optic neuropathy (LHON).
Inherited complex I deficiency can result from mutations in either mitochondrial DNA (mtDNA) or nuclear-encoded structural subunits of the enzyme or from mutation of any of a rapidly expanding number of nuclear-encoded complex I assembly factors. To date, genetic defects have been reported for all seven mtDNA-encoded complex I subunits, 17 of the 38 nuclear-encoded subunits and nine assembly factors. Pathogenic mtDNA mutations may be maternally inherited or sporadic, while most nuclear-encoded complex I defects are inherited as autosomal recessive traits, although a small number of X-linked defects have been reported.
In this review, we discuss the structure, function and assembly of the enzyme; report the findings of a systematic review of the clinical features of 172 published patients with nuclear-encoded complex I defects, including clinical and radiological clues that may aid genetic diagnosis; and consider potential approaches to developing treatments for these devastating disorders.
Structure and function of complex I
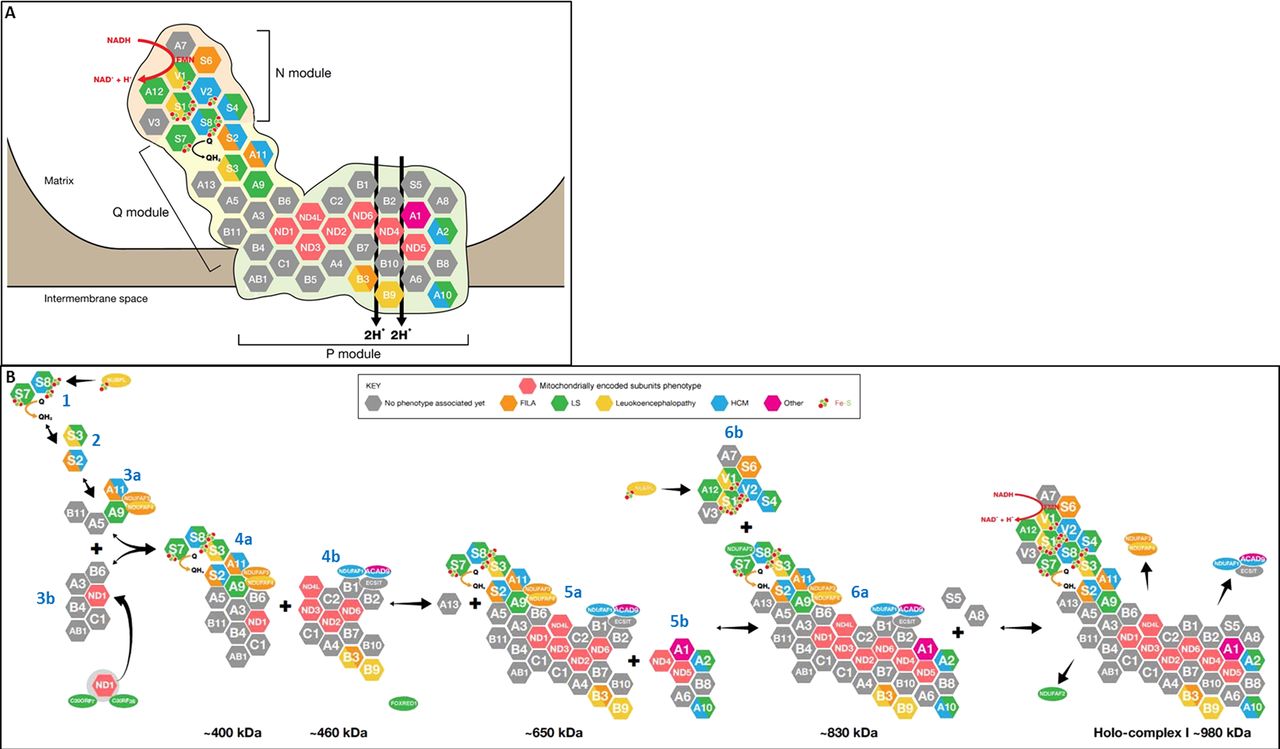
The L-shaped structure of complex I was initially revealed by electron microscopy; further detail was subsequently provided by x-ray crystallography studies of the enzyme in the bacterium Thermus thermophilus and the fungus Yarrowia lipolytica, which demonstrated the relative positions of the subunits in these organisms.2–4 Efforts are underway to determine the positions of the subunits in the mammalian enzyme by crystallising purified complex I from bovine heart. Human complex I is very similar to bovine complex I and consists of 45 different subunits (figure 1A),5 14 of which are necessary for catalytic function and are conserved in all species that have a complex I including bacteria.6 Seven of these ‘core’ subunits are hydrophobic and encoded by mtDNA (ND1, ND2, ND3, ND4, ND4L, ND5 and ND6), whereas the other seven are hydrophilic and encoded by nuclear DNA (NDUFV1, NDUFV2, NDUFS1, NDUFS2, NDUFS3, NDUFS7 and NDUFS8). These 14 subunits have been defined as the ‘minimal enzyme’, while the remaining subunits are often referred to as ‘supernumerary’ or ‘accessory’. The minimal enzyme includes the core subunits of complex I considered essential for catalysing electron transfer from NADH to CoQ10 and generating the proton motive force, as well as the substrate binding sites and all the known redox centres of the enzyme.

Structure and assembly of human mitochondrial respiratory chain complex I. (A) Structure of complex I, showing the 45 subunits (seven encoded by mitochondrial DNA and 38 by nuclear genes), colour-coded according to the clinical phenotype(s) associated with mutations of these genes (see key at the right of figure). Subunits in grey have not yet been linked to human disease. The three functional modules of the enzyme (N electron accepting, Q ubiquinone reducing and P proton pumping) are shown. The oxidation of nicotinamide adenine dinucleotide by flavin mononucleotide generates a flow of electrons that are transported by the Fe–S clusters contained in the subunits NDUFV1-V2-S1-S8 and S7 to ubiquinone, which is consequently reduced to ubiquinol. The energy generated by the electron flow produces a conformational change within the holocomplex which allows for the pumping of four protons (H+) towards the intermembrane space. (B) Assembly pathway of complex I. The main subassemblies (numbered according to the scheme proposed by McKenzie and Ryan7) and the proposed sites of action of the nine assembly factors so far linked to human disease are indicated. Subunits and assembly factors are colour-coded according to the associated phenotype, as shown in the key at the top right.
Complex I has three functional modules: the electron input or N module and the electron output or Q module, both located in the peripheral arm which protrudes into the mitochondrial matrix, and the proton translocase P module within the membrane arm. All seven nuclear-encoded core subunits are located within the N and Q modules, while the seven mtDNA-encoded core subunits are in the P module. Electrons from NADH, which is oxidised at the matrix-protruding end of the peripheral arm, are passed to flavin mononucleotide (FMN), which is non-covalently bound to the NDUFV1 subunit, then via a chain of iron–sulphur (Fe–S) clusters to CoQ10, which is reduced near the junction of the peripheral arm with the membrane arm. The energy generated by the series of electron transfer reactions within the peripheral arm is transduced, by conformational changes in the membrane arm, to pump four protons into the intermembrane space.4 These four protons contribute ∼40% of the electrochemical gradient that drives ATP synthesis.4 The function of the 31 nuclear-encoded supernumerary subunits is still poorly understood, but putative functions include: supporting the structural stability of the enzyme by forming a ‘scaffold’ around the core subunits; protecting the core subunits against oxidative stress; participating in complex I assembly; and regulating the activity of the enzyme.
Complex I assembly
In addition to the structural components of complex I, there are a number of known and putative assembly factors, which chaperone the 45 subunit proteins, one FMN moiety and eight Fe–S clusters through the intricate process of assembling the final ∼980 kDa holoenzyme.7 To date, nine such assembly factors have been linked to human disease (NDUFAF1, NDUFAF2, NDUFAF3, NDUFAF4, C20ORF7, C8ORF38, nucleotide-binding protein-like (NUBPL), FOXRED1 and ACAD9). It is likely that many more complex I assembly factors will be identified considering that the much smaller complex IV, which has only 13 subunits, requires more than 15 assembly factors for its assembly.8 In support of this, phylogenetic profiling studies have identified 25 putative complex I assembly factors.9
Complex I assembly has been studied in various model systems: the fungus Neurospora crassa, mouse cell lines lacking mtDNA-encoded subunits; pulse-chase experiments in human cell lines in which mitochondrial protein synthesis is temporarily blocked by cycloheximide and then allowed to recommence; and in vitro mitochondrial import assays of tagged nuclear-encoded complex I subunits. However, by far the most information about human complex I assembly has come from studies of fibroblasts from patients with mutations in complex I subunits and assembly factors. This has been the subject of intense research interest, which has allowed the identification of at least seven complex I assembly intermediates.7 ,10 So far, precise roles have been elucidated for only a few of the known complex I assembly factors. C20ORF7, C8ORF38, NDUFAF3 and NDUFAF4 have all been implicated early in the complex I assembly process, while NDUFAF1, evolutionary conserved signalling intermediate in Toll pathways (ECSIT) and ACAD9 appear to be involved at an intermediate stage and NDUFAF2 in the late stages.11 Possible functions of the various putative complex I assembly factors/chaperones include assembly of Fe–S clusters, translational coactivation of complex I subunits and direction of nuclear-encoded complex I subunits to the correct intramitochondrial compartment (ie, to the matrix side of the enzyme or to the intermembrane space).7
The first step of complex I assembly is thought to involve incorporation of newly translated mtDNA-encoded subunits into early membrane arm assembly intermediates.10 This step is chaperoned by C20ORF7 which, together with C8ORF38, may function as a translational activator of ND1.12 Alternatively, C20ORF7 may insert ND1 into the membrane or facilitate ND1 into an early membrane arm intermediate. C20ORF7 contains a predicted S-adenosyl methionine-dependent fold, suggesting that it may methylate proteins, RNA or DNA within mitochondria.13 ,14 Only two complex I subunits are known to be methylated:15 NDUFS2 (methylated arginine R323) and NDUFB3 (contains two or three highly conserved methylated histidines).16 Like ND1, NDUFB3 is located in the membrane arm, suggesting that post-translational methylation may play a role in the assembly or stability of the membrane arm. Recently, C20ORF7 mutations were linked to combined deficiency of complexes I and IV,17 and knockdown of C20ORF7 expression in control cells using lentiviral-mediated RNAi18 also led to decreased complex IV activity, suggesting that C20ORF7 may be necessary for assembly of RC supercomplexes.17 ,18
The formation of the peripheral matrix arm begins with the assembly of four core subunits: NDUFS7 and NDUFS8 (intermediate 1), followed by NDUFS3 and NDUFS2 (intermediate 2), to form intermediate 3a (numbering based on nomenclature of McKenzie and Ryan;7 see figure 1B). The assembly factors NDUFAF3 (C3ORF60) and NDUFAF4 (C6ORF66), mutations of which cause fatal neonatal-onset complex I deficiency,19 ,20 tightly associate with intermediate 3a, and it has been suggested that NDUFAF3 and NDUFAF4 may be involved in membrane anchoring of intermediate 2 and promoting maturation to intermediate 3a, which also includes the NDUFA9 subunit (figure 1B). Both NDUFAF3 and NDUFAF4 remain associated with intermediates 4a (∼400 kDa), 5a (∼650 kDa) and 6a (∼830 kDa) as complex I assembly proceeds, but are dissociated just before the formation of the mature holocomplex (figure 1B).19
NDUFAF1 (CIA30) and ECSIT mediate the next step in complex I assembly: the joining of intermediate 4a (a ∼400 kDa subcomplex containing at least NDUFS2, NDUFS3, NDUFS7, NDUFS8 and ND1) with a second membrane arm intermediate 4b of ∼460 kDa (containing at least ND2, ND3 and ND6).10 Pathogenic NDUFAF1 mutations resulted in isolated complex I deficiency and cardiomyopathy in two patients, associated with stalling of complex I assembly at the ∼400 kDa and ∼460 kDa intermediates.21 ,22 No mutations have been identified in ECSIT to date, but mutations in another factor, ACAD9, which also associates with NDUFAF1 and ECSIT, appear to be a relatively common cause of complex I deficiency presenting as HCM and/or exercise intolerance.
As complex I assembly proceeds, NDUFA13 is added to the ∼400 and ∼460 kDa membrane arm intermediates to form intermediate 5a.23 Subunits ND4 and ND5 are then assembled into the growing complex, possibly together with other subunits in the small membrane arm intermediate 5b.24 The resulting ∼830 kDa intermediate 6a remains associated with NDUFAF1 and has been shown by co-immunoprecipitation to contain ND1, ND2, ND3, ND6, NDUFB6, NDUFA9, NDUFS3 and NDUFS7 (but not NDUFS5 or NDUFA8).21 NDUFA1, NDUFA2, NDUFA6,23 NDUFB8 and NDUFA1010 also appear to be assembled into intermediate 6a at this stage. The assembly factor NDUFAF2, mutations of which cause progressive encephalopathy, associates with the ∼830 kDa complex and mediates a late step in the complex I assembly process.25
The last step for completion of fully assembled complex I is insertion of the ∼300 kDa N module (intermediate 6b), which provides the entry point for electrons into the complex.26 In vitro import studies demonstrated the N module to contain at least NDUFS1, NDUFV1, NDUFV2, NDUFV3, NDUFS4, NDUFS6, NDUFA12 and FMN (which is non-covalently bound to NDUFV1).10 Once holocomplex I assembly is complete, the assembly factors NDUFAF1, ECSIT, NDUFAF2, NDUFAF3 and NDUFAF4 dissociate from the mature holoenzyme. The electron transfer activity of complex I also requires the incorporation of eight Fe–S clusters. This step is most likely carried out by at least one assembly factor: HuIND1 (Fe–S protein required for NADH dehydrogenase), also known as NUBPL. NUBPL is a mitochondrial protein which binds Fe–S clusters via a conserved CxxC motif27 and incorporates these into various subunits of the enzyme within intermediates 1 (NDUFS7 and NDUFS8) and 6b (NDUFS1 and NDUFV1).27 ,28
The observation that newly synthesised subunits can be interchanged with pre-existing counterparts within mature complex I suggests that complex I assembly does not always proceed via the linear model described above, but that a subunit exchange mechanism may also be employed to repair damaged enzyme and maintain complex I homeostasis.24 It should be emphasised that the precise mechanism of complex I assembly is still debated, and may be modified as new complex I assembly factors are discovered. A further level of complexity is that it is likely that the majority of complex I exists within RC supercomplexes or ‘respirasomes’ (composed of at least complex I+complex III+complex IV in various stoichiometric ratios).29 Supercomplexes are thought to provide structural and functional advantages to the individual RC enzymes, including stabilisation, protection from degradation, increased efficiency of electron transport and substrate channelling, and decreased electron and proton leakage.30 It has been further suggested that complex I assembly may only occur in the context of the respirasome.31 The existence of supercomplexes may explain the defects of other RC complexes sometimes associated with complex I mutations; for example, complex III defects in some patients with NDUFS4 subunit mutations32 ,33 and complex IV deficiency in occasional patients with defects in the complex I assembly factor C20ORF7.17
Complex I deficiency: clinical phenotypes
Clinical presentation of complex I deficiency is extremely heterogeneous, and ranges from neonatal-onset lactic acidosis to Leigh syndrome and other encephalomyopathies, as well as multisystem disease involvement, and single organ presentations, for example, with HCM or isolated optic neuropathy. The more commonly recognised phenotypes are described below, together with the responsible genes.
Fatal infantile lactic acidosis
The earliest presentation of complex I deficiency is with congenital lactic acidosis, which may present in the neonatal period or early infancy. This disorder is typically rapidly progressive, resulting in death in infancy, and has been linked to mutations in several nuclear-encoded complex I subunits (NDUFV1, NDUFS2, NDUFS6, NDUFS8, NDUFA11 and NDUFB3) and assembly factors (NDUFAF3 and C20ORF7) (see online supplementary table S1). Few mutations have been reported in several of these genes, and so it is difficult to predict whether fatal infantile lactic acidosis (FILA) will be the characteristic clinical presentation for these genetic defects or merely reflects a severe complex I deficiency. An exception seems to apply in the case of NDUFS6 mutations, since these caused FILA in seven children from four unrelated families.34 ,35
Leigh syndrome
The most frequent presentation of complex I deficiency is Leigh syndrome, or subacute necrotising encephalomyelopathy. Affected children typically have normal early development but present in late infancy or early childhood with progressive neurological abnormalities related to brainstem and/or basal ganglia dysfunction. Clinical signs include respiratory abnormalities, nystagmus, ataxia, dystonia and hypotonia. Stepwise neurodevelopmental regression may follow intercurrent illnesses. Often there may be some initial recovery, but never back to the baseline neurodevelopmental trajectory. Leigh syndrome was originally defined neuropathologically (bilateral symmetrical necrotic lesions characterised by the histological quadrad of spongiosis, neuronal loss, astrocytosis and capillary proliferation)36 but now can be diagnosed in life on the basis of the clinical features, elevated lactate levels in blood and/or cerebrospinal fluid and characteristic appearances on MRI of the brain (bilateral symmetrical hyperintensities in the basal ganglia and/or brainstem in T2 weighted sequences).37 Leigh syndrome results from severely impaired cerebral mitochondrial energy production, and is biochemically and genetically extremely heterogeneous. Although any OXPHOS defect may cause Leigh syndrome, isolated complex I deficiency is the most frequently observed biochemical abnormality, accounting for 34% of cases.37 Mutations in six mtDNA-encoded (ND1, ND2, ND3, ND4, ND5 and ND6) and 11 nuclear-encoded (NDUFS1, 2, 3, 4, 7, 8, NDUFV1, NDUFA1, 2, 9, 10) complex I subunits and four assembly factors (NDUFAF2, C8ORF38, C20ORF7 and FOXRED1) have been linked to Leigh syndrome to date (see online supplementary table S1 for details and relevant references).
Leukoencephalopathy
Other patients with infantile-onset complex I deficient encephalomyopathy have a leukodystrophy characterised by cystic white matter changes in the brain MRI. Clinical features in these children include progressive myoclonic epilepsy, episodes of vomiting, global developmental delay and regression, spasticity, dystonia, cerebellar ataxia, ptosis, ophthalmoplegia, nystagmus and optic atrophy. There may be associated macrocephaly. This phenotype has been particularly linked to mutations of two nuclear-encoded complex I subunits, NDUFV1 and NDUFS1, with five and 16 reported cases, respectively (see online supplementary table S1). Neuroradiological appearances in these patients can be confused with vanishing white matter disease.25 ,38–40 A single patient with NDUFS8 mutations also presented with leukoencephalopathy, as did occasional patients with mutations in the NDUFAF4 and NUBPL assembly factors (see online supplementary table S1).
Mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes
Patients with MELAS syndrome usually have symptom-onset in childhood, with seizures, migraines, vomiting, exercise intolerance, proximal limb weakness and short stature. The first stroke-like episode (characterised by transient hemiparesis and/or hemianopsia, often preceded by focal seizures) typically occurs in the first decade of life. Isolated complex I deficiency may be seen in MELAS syndrome, particularly in individuals with ND subunit mutations.41 However, ∼80% of cases are caused by a common mitochondrial transfer RNA (tRNA) mutation m.3243A>G which may also be associated with multiple RC defects. Strokes do not seem to be particularly associated with nuclear-encoded complex I deficiencies, although stroke-like episodes were reported in two Dutch patients with ACAD9 mutations, which are more usually associated with exercise intolerance and/or cardiomyopathy.42
Cardiomyopathy
Complex I deficiency may present in infancy with isolated HCM, sometimes with non-compaction of the left ventricular wall.22 Isolated HCM has been reported with mutations in nuclear-encoded subunits (NDUFS2 and NDUFV240 ,43 ,44) and assembly factors (most commonly ACAD9, usually with associated exercise intolerance,45 but also NDUFAF122). Affected infants may succumb to FILA46 or there may be associated progressive encephalopathy, usually in the Leigh syndrome spectrum, as has been reported for mutations in several genes: NDUFS2, NDUFS4, NDUFS8, NDUFA2, NDUFA10, NDUFA11 and ACAD9 (see online supplementary table S1 for details). It is possible that cardiomyopathy is under-recognised in complex I deficiency, since detailed cardiac investigations may not be performed in some patients with severe neurological presentations. Conduction defects such as Wolff–Parkinson–White syndrome have been reported in patients with both mtDNA (eg, m.13513G>A) and nuclear-encoded (eg, NDUFAF1) complex I defects.21 ,47
Biochemical assessment of complex I
In most centres complex I deficiency is diagnosed by spectrophotometric assay of rotenone-sensitive NADH: ubiquinone oxidoreductase activity in biopsied tissue (usually skeletal muscle, but another affected tissue such as cardiac muscle or liver may be biopsied). Spectrophotometric assays may also be performed in cultured skin fibroblasts from patients, but this is not an ideal tissue for investigation since many patients (particularly those with mtDNA mutations, but also some nuclear-encoded defects) do not express complex I deficiency in fibroblasts. In most specialist centres, fibroblasts are assayed as a second-line investigation in order to determine whether there is a systemic complex I deficiency or a tissue-specific defect. Rotenone is used in the assay because there are many NADH oxidoreductases in the cell, but only complex I is rotenone-sensitive; residual NADH oxidoreductase activity after rotenone administration is subtracted from total NADH oxidoreductase activity in order to derive complex I activity. The activity of complex I is usually expressed as a ratio to a mitochondrial matrix enzyme (most commonly one of the Krebs cycle enzymes, such as citrate synthase or succinate dehydrogenase) to control for varying mitochondrial content between samples.
The biochemical diagnosis of complex I deficiency is not trivial and consequently there are no universally accepted diagnostic criteria. Measurement methods and reference ranges vary between laboratories: some centres use values <30% of the control mean; others use anything below the control range; and quality assurance schemes are still in their infancy.48 Isolated complex I deficiency refers to a severe reduction of complex I, with activities of other OXPHOS complexes within (or close to) the reference range.1 A potential caveat is that the spectrophotometric NADH to ubiquinone oxidoreductase assay is only a measure of redox activity within the peripheral arm. Mutations of membrane arm subunits, which affect proton pumping rather than electron transfer, may theoretically result in apparently ‘normal’ enzyme activity. Therefore, normal complex I enzyme activity does not completely exclude the possibility of complex I deficiency, as has been demonstrated for patients with mutations in the peripherally located ND5 subunit.41 More recently mini-oxygraphy methods have been developed; while these do not give a specific measurement of complex I activity, they do allow assessment of the global oxygen consumption capacity of isolated mitochondria or intact cells.49 ,50 In addition, immunocapture-based methods have been used to interrogate the function of complex I.22 ,51 The presence of individual complex I subunits may be determined by western blot analysis, and the technique of blue native gel electrophoresis (BNGE) is proving to be a powerful method for studying the assembly of the complex I holoenzyme and dissecting specific complex I assembly defects.10
Genetics of complex I deficiency
Complex I deficiency is genetically extremely heterogeneous and several patterns of inheritance have been observed, including maternal, autosomal recessive and X-linked. This genetic complexity, superimposed on the clinical heterogeneity discussed above, leads to considerable difficulties in establishing genetic diagnoses for patients with complex I deficiency. Complex I disease genes have been identified by candidate gene analysis (NDUFAF1,21 NDUFAF2,25 C8ORF389); genetic linkage and homozygosity mapping approaches (NDUFAF3,19 NDUFAF4,20 C20ORF7,18 FOXRED151); and more recently by targeted or whole exome sequencing using next generation sequencing (NGS) techniques (NUBPL,52 ACAD953).
mtDNA mutations
Maternally inherited mutations in mtDNA-encoded complex I subunits (ND subunits) were initially linked to LHON almost 25 years ago.54 Mutations associated with LHON are homoplasmic. Subsequently, heteroplasmic mutations in ND subunits were reported in association with other clinical phenotypes, such as dystonia, MELAS and Leigh syndrome. Several recurrent mutations are recognised,55–58 and the ND5 subunit appears to be a particular hotspot for disease-causing mutations.41 Many patients have features overlapping different mitochondrial syndromes, for example some patients have LHON plus dystonia and other patients may have features overlapping MELAS, LHON and Leigh syndrome.41 A complete list of the mtDNA mutations associated with complex I deficiency can be found in the online Mitomap database (http://www.mitomap.org/MITOMAP). Mutation pathogenicity can be especially difficult to prove for mtDNA mutations since mtDNA is extremely polymorphic and many mutations are ‘private’ to individual families. Factors supporting pathogenicity include heteroplasmy, segregation with disease within a family, association with a similar disease phenotype in multiple unrelated families and (the gold standard, but only possible in cases where the biochemical defect is expressed in cultured cells) demonstration of transfer of the biochemical phenotype with the mtDNA mutation in transmitochondrial cybrids. Mutation pathogenicity scoring criteria can also be helpful.59 It should be noted that many mtDNA mutations are sporadic and so the absence of a family history suggestive of maternal inheritance does not exclude the possibility of a mtDNA mutation. Studies of several cohorts of patients with complex I deficiency from around the world have suggested a fairly uniform prevalence of causative mtDNA mutations of ∼20%–30%.58 ,60 ,61 These studies indicate that sequencing mtDNA is a useful first-line genetic screening strategy in complex I deficiency, since it will be possible to make a genetic diagnosis in a significant minority; however, most cases will have a nuclear defect.
Nuclear subunit mutations
So far, mutations in 17 of the 38 nuclear-encoded complex I subunits have been reported to cause complex I deficiency, including all seven nuclear-encoded core subunits and 10 of the supernumerary subunits (see online supplementary table S1). The first nuclear subunit mutations were identified by systematically sequencing core subunit genes in a relatively large cohort of patients,62–65 and subsequently other mutations were identified by homozygosity mapping approaches in consanguineous families.18 ,35 To date, more than 100 affected patients have been reported: ∼60% with core subunit mutations and ∼40% with mutations in accessory subunits (see online supplementary table S1). The most frequently observed phenotypes were Leigh syndrome, leukoencephalopathy and HCM (see online supplementary table S1; figure 1A and figure 2A). Most mutations were reported for NDUFS1 (24 cases) and NDUFS4 (21 cases), but mutations of NDUFV1 and NDUFS2 also occurred relatively frequently (14 and 12 cases, respectively). It is not yet known whether the preponderance of core subunit mutations reflects ascertainment bias (these subunits are more likely to be included in targeted candidate gene screening projects) or whether core subunit mutations are more likely to have functional consequences and that mild mutations in accessory subunits can be tolerated without causing clinical disease. Large-scale exome sequencing projects, which are not subject to ascertainment bias, should help to answer this question.

Genotype to phenotype correlations in nuclear-encoded complex I deficiency. (A) Venn diagram illustrating genotype to phenotype correlations between mutations in nuclear-encoded complex I subunits and assembly factors and the main clinical phenotypes (neurological, metabolic, cardiac and exercise intolerance). Note the considerable genetic heterogeneity for each clinical subgroup, and that several genetic defects are associated with more than one phenotype. (B) Kaplan–Meier survival curves for nuclear-encoded complex I deficiency, according to age of disease onset. All survival functions were calculated using SPSS V.20. (C) Pie chart illustrating the relative prevalence of the main clinical phenotypes of nuclear-encoded complex I deficiency. (D) Kaplan–Meier survival curves for nuclear-encoded complex I deficiency, according to clinical phenotype. (E) Kaplan–Meier survival curves for defects in nuclear-encoded complex I subunits, compared with assembly factor defects. (F) Blood and cerebrospinal fluid lactate concentrations (mM) reported in patients with mutations in nuclear-encoded subunits (red) and assembly factors (blue) of complex I. Normal lactate concentration is <2 mM.
Assembly factor mutations
Nine nuclear-encoded complex I assembly factors have now been linked to human disease, with mutations reported in 55 patients from 32 families so far (see online supplementary table S1). The first complex I assembly defect was found using a ‘genome subtraction’ method, in which genes that were present in fungi with complex I but absent in fungal species without complex I were identified as putative complex I assembly factors. This approach led to the discovery of B17.2L (now renamed NDUFAF2) mutations in a patient with progressive encephalopathy.25 Eight further patients with NDUFAF2 mutations have since been reported (see online supplementary table S1), all with similar clinical features and a characteristic neuro-imaging appearance (see below). Mutations in NDUFAF1 were linked to HCM in two unrelated patients by BNGE profiling and candidate gene sequence analysis,21 ,22 while homozygosity mapping revealed NDUFAF3 and NDUFAF4 to be complex I assembly factors associated with FILA or severe infantile-onset encephalopathies (see online supplementary table S1). Phylogenetic profiling identified C8ORF38, C20ORF7 and FOXRED1 as candidate complex I assembly factors, and these were all subsequently linked to human disease using homozygosity mapping.9 ,18 ,51 Finally, mutations in NUBPL were identified using a targeted next generation sequencing approach,52 while mutations in ACAD9 (previously thought to be involved in fatty acid oxidation) were discovered to cause complex I deficiency associated with HCM and/or exercise intolerance in a whole exome sequencing project.53
Other genetic causes of complex I deficiency
Because complex I contains the largest number of mtDNA-encoded subunits, isolated complex I deficiency may be the initial biochemical defect in disorders of mtDNA replication and translation, although later in the course of these disorders there are usually multiple RC defects. The most frequent cause of defective mtDNA replication is mutation of the POLG gene encoding the catalytic subunit of DNA polymerase γ, and occasional patients with POLG mutations do present with isolated complex I deficiency. The most frequently diagnosed mtDNA translation defects affect the mitochondrial tRNA molecules, either as point mutations involving a single tRNA or large-scale rearrangements which may delete several tRNA genes. More recently, nuclear-encoded defects of mitochondrial translation have been linked to isolated complex I deficiency; for example, mutations in the MTFMT gene encoding the mitochondrial methionyl-tRNA formyltransferase.66
Approaches to diagnosis
From a clinical viewpoint, the important questions in complex I deficiency are whether there are characteristic clinical features of complex I deficiency; whether any particular clinical features should arouse suspicion of specific gene defects; and finally whether specific gene defects are associated with a better or worse prognosis. Traditionally, it has been very difficult to identify genotype to phenotype correlations for mitochondrial disorders, including complex I deficiency, because of the extreme genetic heterogeneity underlying these diseases. Furthermore, even the most specialised centres will only see small numbers of patients with particular genetic defects, and so it is difficult for physicians to identify clinical clues that may point to specific diagnoses. A further source of bias is the referral pattern for individual clinicians; for example, different subgroups of patients are likely to be referred to neurologists compared with metabolic physicians or biochemical geneticists.
We sought to address these difficulties by performing a systematic review of all cases of genetically confirmed nuclear-encoded complex I deficiency in order to search for genotype to phenotype correlations and identify clinical, radiological or biochemical patterns that may help to expedite genetic diagnosis for affected individuals. Extensive literature searches of the PubMed database were performed by both authors, using the key words complex I deficiency, and names and aliases of all the nuclear-encoded complex I subunits and known assembly factors in order to obtain as near complete an ascertainment as possible of all cases published in the 14-year period, February 1998–April 2012 inclusive. All cases of genetically confirmed complex I deficiency with nuclear mutations were included in the review. Cases where only one mutation had been identified were excluded, with the exception of five hemizygous males and a single heterozygous female with mutations in the X-linked NDUFA1 gene. Other exclusion criteria were apparent duplicate reports and cases where a mutation was reported but no clinical information was supplied. In all, 65 papers were included in the review, reporting a total of 172 patients: 117 with nuclear subunit mutations and 55 with assembly factor mutations. A full list of the publications included in our meta-analysis is given in the online supplementary material.
Genotype to phenotype correlations
The overall male to female ratio observed was 1.4 : 1 but when this was broken down according to subtype of genetic defect, the proportion was 1.7 : 1 for nuclear subunit mutations and 1 : 1 for assembly factor mutations. The reason for the male preponderance with nuclear subunit mutations is not clear, since only a handful of cases had mutations in the X-linked NDUFA1 gene. Approximately 30% of cases had symptom-onset in the neonatal period or infancy and a further ∼60% in early childhood, meaning that the overwhelming majority of cases with nuclear-encoded complex I defects present before 5 years of age. The distribution of age of onset was roughly the same for subgroups with nuclear subunit mutations and assembly factor defects (data not shown). In general, survival and rates of disease progression are broadly related to age at onset (figure 2B). The largest subgroup of patients presented with early-onset neurodegenerative disease with symptoms/signs compatible with the Leigh syndrome spectrum (39% of cases). These patients had mutations in 21 different genes, so it is difficult to deduce genotype to phenotype correlations for complex I deficiency with Leigh syndrome/Leigh-like features (see online supplementary table S1; figure 1). Other patients presented with a leukoencephalopathy (14%), an unspecified encephalomyopathy (9%) or FILA (11%). Overall, 19% of cases had HCM, associated with Leigh syndrome or other encephalopathic illness in over half of these cases. The remaining 8% of cases had miscellaneous clinical features, including exercise intolerance (4%), myoclonic epilepsy (2%), cerebellar ataxia (2%) and recurrent lactic acidosis in a single case (figure 2C). By definition, survival was poorest in those with FILA, while those with isolated exercise intolerance had the best survival. Rates of progression and survival were broadly the same for all other phenotypic subgroups (figure 2D). Overall survival appeared to be longer for patients with assembly factor mutations, which is largely attributable to patients with ACAD9 mutations and exercise intolerance (figure 2E).
Defects in most genes (except where only a single case has been reported) are associated with considerable clinical heterogeneity, as illustrated in figure 2A. For example, mutations in NDUFAF1 led to fatal infantile HCM in one patient,22 but an initially severe HCM later improved in another patient who was still alive at 20 years.21 Similarly, the clinical spectrum associated with mutations in C20ORF7 ranges from neonatal-onset mitochondrial disease leading to death within a few days to adults with relatively mild Leigh syndrome associated with survival into the fourth decade.18 ,42 The factors contributing to this observed clinical variability are not well understood, but possible explanations include genetic modifiers, environmental factors (eg, exposure to severe viral illnesses, surgery and other metabolic stresses) and modulation of phenotype by altered immune signalling.22 A notable exception to this lack of genotype to phenotype correlation is the case of ACAD9 mutations, where nearly all reported cases had HCM and/or exercise intolerance.45 ,53 ,67 These patients were also characterised by clinical response to riboflavin supplementation.67 However, given the small number of patients reported with mutations in most of the complex I nuclear subunit and assembly factor genes, it is difficult to draw definitive conclusions about genotype to phenotype correlations.
Neuroimaging clues
MRI brain changes are frequently observed in patients with complex I deficiency, but in most cases are neither specific nor associated with particular genetic defects. A single-centre retrospective review of MRI scans from 30 patients with genetically confirmed complex I deficiency revealed involvement of brainstem structures in 100% of their patients and basal ganglia lesions (particularly affecting the putamina) in 90%.68 The brainstem lesions appeared as hyperintensities in the T2 and Fluid Attenuated Inversion Recovery (FLAIR) sequences and were hypointense in T1-weighted images. Within this series, stroke-like lesions appeared to associate with mtDNA mutations and leukoencephalopathy with nuclear subunit mutations. Cerebellar involvement was noted in ∼45% of cases, and occurred with both mtDNA and nuclear gene defects.68 Lebre et al reported that the combination of brainstem and striatal lesions was infrequently observed in their control groups (MT-TL1 mutations and pyruvate dehydrogenase deficiency), but comparison was not made with other RC defects causing Leigh syndrome (eg, complex IV deficiency caused by SURF1 mutations and complex V deficiency caused by MT-ATP6 mutations), which may present with similar MRI appearances.37
In our systematic review, detailed MRI brain reports were available for 82 of the 172 patients with nuclear-encoded complex I deficiency. Of these, only 13% had isolated basal ganglia lesions, while 28% had isolated brainstem lesions and 24% had both basal ganglia and brainstem lesions, supporting the notion that brainstem lesions may be particularly frequent in complex I deficient Leigh syndrome. However, systematic studies of other causes of Leigh syndrome are needed to determine the specificity of this observation, as discussed in the previous paragraph. A highly specific neuroimaging pattern was only seen with mutations in the NDUFAF2 assembly factor: brainstem lesions within the mamillothalamic tracts, substantia nigra, medial lemniscus, medial longitudinal fasciculus and spinothalamic tracts on T2-weighted scans.25 ,52 ,69–71 These patients did not have changes in the thalami and basal ganglia. In all, 24% of all complex I deficient cases in our review had neuroimaging features of leukoencephalopathy, most frequently associated with NDUFS1 (16 cases) and NDUFV1 (five) mutations, but also in single cases with NDUFS8, NDUFAF3 and NUBPL mutations. Cerebellar involvement was reported in nine cases, spinal cord lesions were documented in three cases and four patients had partial or complete agenesis of the corpus callosum. It is possible that other specific imaging patterns may emerge for subgroups of complex I deficiency, as further patients are genetically characterised. For now, MRI appearances of Leigh syndrome with brainstem and basal ganglia involvement cannot be considered sufficiently specific to avoid the need for muscle biopsy and determination of specific RC enzyme activities.
Histological clues
Most children with complex I deficiency have only minor nonspecific abnormalities in muscle histology, for example, mild lipid accumulation or fibre type disproportion. The presence of ragged red fibres should arouse suspicion of an underlying mtDNA defect, which may be a large-scale rearrangement or point mutation, or a defect of mtDNA maintenance or translation. Ragged red fibres are not usually observed in nuclear-encoded complex I defects but were reported in single cases with NDUFS4, NDUFS7, FOXRED1 and NUBPL mutations.32 ,52 ,72 In addition, occasional patients with nemaline rods and complex I deficiency have been reported,73 and in one of these cases mutations of the structural subunit NDUFB3 were identified recently.74
Biochemical clues
Plasma and cerebrospinal fluid lactate were frequently elevated in the reported cases and do not appear to discriminate between different molecular genetic defects, nor was there any significant difference between patients with nuclear subunit mutations and those with assembly factor defects (figure 2F). Moreover, lactate levels did not correlate with residual complex I activity. The results of other metabolic investigations (plasma amino acid and acylcarnitine profiles and urinary organic acids) were very infrequently reported, and so it is not possible to draw any conclusions regarding whether these might provide diagnostic clues towards specific molecular genetic defects. As expected, most cases had isolated deficiency of complex I in skeletal muscle and fibroblasts (and other tissues such as heart and liver where they were assayed). In most cases, residual activity of complex I was greater in fibroblasts than in skeletal muscle. There does not appear to be any correlation between residual enzyme activity and specific genetic defect (see online supplementary table S1). In occasional cases, a more widespread OXPHOS defect was observed; for example, a recent report described complexes I and IV deficiencies in a family with C20ORF7 mutations.17 Similarly, five patients with NDUFS4 mutations were reported to have combined deficiencies of complexes I and III32 ,62 ,75 ,76 and a further two cases had a combined defect of complexes I and IV.52 However, the majority of patients with mutations in these genes had isolated complex I deficiency. Possible explanations for the presence of multiple OXPHOS deficiencies in patients with complex I mutations are that increased ROS generated by dysfunctional complex I cause oxidative damage to other OXPHOS enzyme complexes or that mutation of particular complex I subunits leads to instability of RC supercomplexes, with subsequent degradation and therefore loss of activity of enzymes not assembled into supercomplexes.
Analysis of complex I assembly in patient tissues by BNGE is emerging as a method for identifying patients with abnormal subassemblies of the enzyme, and directing genetic investigations towards particular candidate genes in these patients. For example, all patients with NDUFS4 mutations reported in the literature accumulate a ∼830 kDa subassembly lacking the N module;77 BNGE screening may be the most efficient way to detect this subgroup of patients. Patients with NDUFAF1 defects also appear to have a characteristic subassembly profile, with accumulation of the ∼400 and ∼460 KDa subassemblies.21 ,22
On the basis of the above clinical, neuroradiological, biochemical and genetic considerations, we suggest a diagnostic flowchart as depicted in figure 3.

{kind=link}
{kind=link}
{kind=link}
Diagnostic flowchart for isolated complex I deficiency. Proposed pathway for genetic investigations in patients with isolated complex I deficiency. *For suggested diagnostic pathway for other oxidative phosphorylation defects, see the review by Rahman and Hanna.99 **This step is optional; because of the large number of potential candidate genes it may be preferable (and more cost-effective) to move to straight to whole exome next generation sequencing (NGS), whilst accepting the considerable bioinformatics challenges inherent to whole exome sequence analysis. See figure 2A for candidate genes associated with neurological, metabolic or cardiac presentations of isolated complex I deficiency. mtDNA, mitochondrial DNA. NB nota bene.
Secondary complex I deficiency
A number of ‘secondary’ causes of complex I deficiency have been reported, most notably Parkinson's disease (PD).78 Complex I deficiency was first linked to PD in the 1980s when it became apparent that there was a high incidence of PD in people who had recreationally used MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), a known inhibitor of complex I.79 Subsequently, a specific reduction of complex I activity was noted in postmortem substantia nigra specimens from subjects with PD.80 Mutations of mtDNA-encoded subunits of complex I have been associated with various cancers, notably thyroid oncocytic tumours.81 Furthermore, we have recently shown that an MT-ND2 mutation appears to be involved in tumour cell resistance to the chemotherapeutic agent cisplatin.82 Complex I deficiency has also been linked to several other disease processes, including autism,83 ,84 diabetes mellitus85 and a subtype of Charcot Marie Tooth disease.86 A detailed discussion of secondary complex I deficiencies is not possible here owing to space constraints; the reader is referred to a recent review by Schapira.78
Pathogenic mechanisms
While impaired ATP production is undoubtedly a major consequence of complex I deficiency, effects of mutations on the other functions of complex I are also likely to play a significant part in the pathogenesis of clinical disease. For example, complex I is a major site of ROS production and ROS are now regarded as important signalling molecules effecting communication between mitochondria and other subcellular compartments. Studies have shown that superoxide production is inversely correlated with complex I activity in complex I deficient fibroblasts.87 Furthermore, fibroblasts with very low residual activity had increased levels of ROS and fragmented mitochondrial morphology,88 suggesting that these deficient mitochondria were being targeted for autophagocytic destruction or mitophagy.89 The membrane potential is reduced in complex I deficient fibroblasts90 ,91 and there appears to be a linear correlation between membrane potential and increased superoxide-derived ROS levels.92 Finally, reduced ATP production was closely related to ROS levels and membrane potential,93 suggesting that all of these factors are likely to play a cumulative role in mediating disease pathogenesis.
Mouse models of complex I deficiency
The recent development of a number of mouse models of complex I deficiency is likely to lead to advances in understanding disease mechanisms in complex I deficiency. The first mutant mouse reported to have complex I deficiency was the Harlequin mouse, which has a hypomorphic mutation in the Aif gene encoding the apoptosis-inducing factor.94 However, although the mutant mice appear to have isolated complex I deficiency, mutations in the human homologue AIFM1 have been reported to cause progressive encephalomyopathy with multiple RC defects rather than isolated complex I deficiency,95 and so the Harlequin mouse may not be the best model for human complex I deficiency. Since many knockout mouse models of nuclear-encoded mitochondrial genes are embryonic lethal, mice with a conditional deletion of Ndufs4 exon 2 were created using the Cre/loxP recombination system. These mice are born apparently healthy but develop ataxia from 5 weeks, leading to death from progressive encephalomyopathy by 7 weeks.96 Moreover, mice with conditional knockout of Ndufs4 in the central nervous system have neuropathological features resembling Leigh syndrome.97 The Ndufs6 gene trap mouse model has isolated complex I deficiency manifesting as cardiomyopathy starting from postnatal day 30, with heart failure and weight loss, sometimes causing sudden death, at approximately 4 months in the male mice and 8 months in the female animals.98 Residual complex I activity is ∼10% of control values, reflecting very low levels of fully assembled enzyme, and ATP production is severely reduced in isolated mitochondria using substrates needing complex I for oxidation. ROS production appears to be normal in these mutant mice. Although none of these mouse models perfectly replicates human disease (eg, human NDUFS6 mutations have never been reported to cause HCM), it is anticipated that these animal models will prove to be invaluable tools in unravelling pathogenic mechanisms underlying mitochondrial disease, as well as providing a platform for preclinical trials of candidate therapies for complex I deficiency.
Approaches to treatment
Disappointingly, it is still the case that there are no effective curative therapies for the majority of cases of complex I deficiency, and symptomatic measures remain the mainstay of treatment for most patients.99 However, it has been known for almost 20 years that occasional patients, particularly those with a myopathic presentation, may show a clinical response to supplementation with the vitamin riboflavin (B2)100 ,101. Until recently, most of these patients did not have a genetic diagnosis, with the exception of a case with a complex I deficient myopathy caused by the m.3250T>C mtDNA mutation.102 Riboflavin is necessary for the synthesis of FMN and flavin adenine dinucleotide. Complex I contains a single FMN moiety, non-covalently bound to the NDUFV1 subunit, but patients with NDUFV1 mutations do not appear to be particularly responsive to riboflavin. However, a number of recent reports have demonstrated that riboflavin responsiveness in nuclear-encoded complex I deficiency is related to ACAD9 deficiency. ACAD9 is a flavoprotein-containing enzyme which was initially implicated in long chain fatty acid oxidation103 but now appears to have a more convincing role in complex I assembly.45 ,53 ,67 Increased flavin adenine dinucleotide availability as a result of riboflavin supplementation is thought to stabilise mutant flavoproteins and thereby increase their activity.104 Riboflavin supplementation was shown to increase complex I activity approximately twofold in cultured fibroblasts bearing ACAD9 mutations,53 and residual muscle complex I activity also increased from 16% to 47% in a repeat biopsy taken 2 years after riboflavin therapy was commenced in one man.105 Riboflavin treatment (at doses ranging from 50 mg/day in a neonate to 100–300 mg/day in adults) has been documented for five patients with ACAD9 mutations, and symptomatic improvement was reported in all cases, who were alive aged 5–24 years at the time of the reports.53 ,67 ,105 However, six patients with ACAD9 mutations reported in the literature died, between the ages of <1 month and 12 years.45 ,53 ,74 It is not clear from the reports whether any of these children received riboflavin.
Although a therapeutic trial of riboflavin should be mandatory for all patients with complex I deficiency, most patients are unlikely to respond. There is a clear need for other treatment strategies. The most promising approaches involve antioxidant compounds or target mitochondrial biogenesis. The role of the antioxidant vitamin E and its analogues such as Trolox in complex I deficiency was the subject of a recent review.106 Analogues of another antioxidant, coenzyme Q10, have shown promise in arresting disease progression in LHON if given early.107 ,108 Mitochondrial biogenesis may be stimulated via various pharmacological agents, which all appear to act via the common pathway of stimulating the PGC1α translational coactivator.109 Bezafibrate and AICAR (5-amino-1-β-D-ribofuranosyl-imidazole-4-carboxamide), which both stimulate PGC1α, were recently shown to improve various measures of mitochondrial function in cultured skin fibroblasts from patients with nuclear-encoded complex I deficiency.110 The ketogenic diet has also been proposed to increase mitochondrial biogenesis,111 and there are anecdotal reports of benefit from a ketogenic diet in occasional patients with complex I deficiency; for example, temporary improvement of ptosis and ophthalmoplegia in a child with NDUFV1 mutations.112 However, other reports have suggested that increasing dietary fat does not improve complex I deficiency.113 Finally, two recent reports of successful gene therapy in rat models of LHON offer hope for patients with this subgroup of complex I deficiency.114 ,115
While the above strategies all show promise, most have not yet reached the stage of preclinical trials, and much work remains to be done in devising effective therapies for complex I deficiency. However, although developing new more effective treatments is undoubtedly important, we must not lose sight of the fact that there is an urgent need for well designed and adequately powered clinical trials of the most promising agents sooner rather than later.
Conclusions
Complex I deficiency is a common cause of childhood-onset mitochondrial disease, but the associated clinical and genetic heterogeneity leads to considerable diagnostic challenges. Recent advances in genetic techniques, particularly the availability of relatively inexpensive high-throughput whole exome next generation sequence analysis, have led to the identification of the causative gene in large numbers of patients in the last 2 years. This has allowed some tentative genotype to phenotype correlations to be made. For example, patients with ACAD9 mutations typically have HCM and/or exercise intolerance, while those with NDUFAF2 defects have a subtype of Leigh syndrome with a highly specific neuroimaging appearance. These emerging genotype to phenotype correlations are important, since they will allow the diagnostic process to be more rapid, which is crucial for affected families seeking genetic counselling and prenatal diagnosis. However, some phenotypes, notably Leigh syndrome, are characterised by extreme genetic heterogeneity, and the numbers of reported patients with mutations in many of the causative genes are too small to allow genotype to phenotype comparisons to be made. An ongoing challenge is that a molecular diagnosis remains elusive for approximately 50% of patients with complex I deficiency, despite high-throughput sequencing.9 ,74 There are several possible explanations for this: determining which of the many variants identified by exome sequencing is pathogenic is a huge bioinformatic task; mutations may lie in introns or untranslated regulatory regions, and the assumed inheritance pattern may not be correct (eg, some patients may have de novo dominant mutations, rather than recessive mutations as is usually assumed for severe early-onset mitochondrial diseases in which mtDNA mutations have been excluded). Finally, identification of complex I deficiency should prompt initiation of riboflavin treatment since some patients, particularly those with ACAD9 mutations, may respond to supplementation with this vitamin. However, effective treatments are still lacking for the majority of patients with this devastating group of disorders. The recent development of several mouse models will be invaluable for preclinical trials of candidate therapies, but much work remains to be done.
Acknowledgments
EF and SR are supported by Great Ormond Street Hospital Children's Charity.
References
Supplementary materials
Article with updated figures
Files in this Data Supplement:
- final article PDF
- Data supplement 1 - Supplement 1
Footnotes
-
Contributors Both authors performed a systematic review of the literature analysed the data, and SR wrote the manuscript.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Miscellaneous