Article Text

Abstract
Background Next generation sequencing has become the core technology for gene discovery in rare inherited disorders. However, the interpretation of the numerous sequence variants identified remains challenging. We assessed the application of exome sequencing for diagnostics in complex I deficiency, a disease with vast genetic heterogeneity.
Methods Ten unrelated individuals with complex I deficiency were selected for exome sequencing and sequential bioinformatic filtering. Cellular rescue experiments were performed to verify pathogenicity of novel disease alleles.
Results The first filter criterion was ‘Presence of known pathogenic complex I deficiency variants’. This revealed homozygous mutations in NDUFS3 and ACAD9 in two individuals. A second criterion was ‘Presence of two novel potentially pathogenic variants in a structural gene of complex I’, which discovered rare variants in NDUFS8 in two unrelated individuals and in NDUFB3 in a third. Expression of wild-type cDNA in mutant cell lines rescued complex I activity and assembly, thus providing a functional validation of their pathogenicity. Using the third criterion ‘Presence of two potentially pathogenic variants in a gene encoding a mitochondrial protein’, loss-of-function mutations in MTFMT were discovered in two patients. In three patients the molecular genetic correlate remained unclear and follow-up analysis is ongoing.
Conclusion Appropriate in silico filtering of exome sequencing data, coupled with functional validation of new disease alleles, is effective in rapidly identifying disease-causative variants in known and new complex I associated disease genes.
- Exome sequencing
- mitochondrial complex I deficiency
- NDUFB3
- MTFMT
Statistics from Altmetric.com
Introduction
The advent of next generation sequencing (NGS) technology facilitates the discovery of new disease genes and is expected to transform and complement routine clinical diagnosis. This is particularly true for conditions characterised by extensive genetic heterogeneity that hampers the prioritisation of obvious candidate genes and the identification of causative mutations.
Human mitochondrial respiratory chain complex I deficiency (MIM 252010) is the most common biochemical signature of mitochondrial disorders, a group of highly heterogeneous conditions characterised by faulty oxidative phosphorylation (OXPHOS), which collectively affects 1 in 5–10 000 live births.1 Human complex I is a giant multiheteromeric structure of 45 subunits. Among these are eight Fe-S clusters and an FMN moiety, which transfer electrons from NADH to ubiquinone, using the free energy of this process to pump protons across the inner mitochondrial membrane. Seven subunits of complex I are encoded by mtDNA genes, the remaining ones by nuclear genes. Several additional gene products have been shown to contribute to its formation and activity, and further ones are hypothesised.2 This structural and functional complexity can explain, at least in part, the extraordinary heterogeneity of complex I defects that may range from mild to severe. Complex I deficiency is known to be associated with a broad spectrum of clinical presentations, affecting single or multiple organs at any time in life.3 Obvious genotype-phenotype correlations are missing, making inference from the biochemical or clinical presentation on the gene involved difficult or impossible.4 Complex I deficiency is caused by a large set of disease genes, most of which are still unknown. In fact, mutations associated with complex I deficiency have been reported in 21 complex I subunits encoded by the seven mtDNA genes, and by 17 nuclear genes and in 10 accessory factors essential for complex I assembly and stability. However, these genes account for only half of the biochemically defined cases of complex I deficiency.5 6 Taken together, these considerations make complex I deficiency a paradigmatic disorder to validate NGS protocols for diagnostic purposes.
A major hurdle for NGS to become the standard approach in routine molecular screening is the difficulty to distinguish between causal mutations and thousands of DNA sequence variants present in each individual. In this context, a distinct advantage of the biochemically well defined OXPHOS disorders is the possibility to validate the pathogenicity of a given variant by integrating in silico analysis with functional complementation in mutant cells (figure 1).

Schematic overview of the exome study in complex I deficiency. CI, complex I; NSV, non-synonymous variants. Mito-genes refers to genes coding for known and predicted (MitoP2 SVM score ≥1.0) mitochondrial proteins. Numbers in parentheses indicate estimated numbers for each category.
Patients, materials and methods
Patients
Informed written consent was obtained from all participants or their guardians, and the ethics committee of the Technical University of Munich approved the study. We evaluated 10 index patients with isolated complex I deficiency. This diagnosis was based on established diagnostic criteria7 and disease manifestation was before the age of 3 years. Accordingly, seven patients (33027, 37796, 44409, 46072, 50845, 59029, 61606) had residual complex I activities of <25% of the mean of ≥10 healthy controls. Another three patients (33281, 33284, 33463) had residual complex I activities between 26% and 40% in muscle and fibroblasts. Complex II, III, and IV deficiencies were biochemically excluded in all patients and none of the patients had a molecular diagnosis before. Rare mtDNA variants had been excluded by Sanger sequencing of the entire mtDNA. The clinical presentation and biochemical findings of all patients and affected siblings are summarised in table 1. A detailed case report of patient 50845 with causative mutations identified in NDUFB3 is provided in the supplementary data.
Clinical and biochemical features of index patients and affected siblings
Exome sequencing and variant filtering
Within the framework of the German network for rare diseases ‘mitoNET’ and the European E-Rare program ‘GENOMIT’ we established an exome sequencing and variant filtering pipeline for the molecular diagnosis of mitochondrial disorders. The exome of individual 33284 was sequenced as 54 bp paired-end runs using two lanes of a flowcell on a Genome Analyser IIx system (Illumina, San Diego, CA, USA) after in-solution enrichment of exonic sequences using the SureSelect Human All Exon 38 Mb kit (Agilent, Santa Clara, CA, USA). All other samples were processed with the SureSelect Human All Exon 50 Mb kit (Agilent) and subsequently sequenced as 76 bp paired-end runs to an average 120 × coverage corresponding to 9–12 Gb of sequence data. Read alignment was performed with BWA (version 0.5.8 or 0.5.9) to the human genome assembly hg19. Single nucleotide variants and small insertions and deletions were detected with SAMtools (V.0.1.7). Since mitochondrial disorders are rare conditions, we excluded variants present with a frequency >0.4% in 879 control exomes. Assuming an autosomal recessive mode of inheritance, homozygous or compound heterozygous variants were searched for (table 2), yielding a median of 28 mutated genes per individual.
Variants identified in 10 individuals with complex I deficiency by exome sequencing
We then applied three sequential filters to identify the causal mutations: (1) a first filter for disease variants known to be associated with respiratory chain defects according to the Human Gene Mutation Database; (2) a second filter based on the identification of novel homozygous or compound heterozygous variants affecting genes encoding complex I subunits or known complex I assembly factors; and (3) a third filter for genes encoding known and predicted mitochondrial proteins listed in MitoP2.8
Rescue experiments and investigation of complex I assembly
In order to prove the pathogenicity of newly identified missense alleles, we tested the biochemical rescue of complex I defective skin fibroblast cell lines from three index cases after lentiviral mediated expression of wild type (wt) cDNAs using the p.Lenti6.3/V5-TOPO vector system (Invitrogen).9 Complex I activity was measured as rotenone-sensitive oxygen consumption rate using the Seahorse system (Bioscience, North Billerica, MA, USA).10
In the same samples, we also analysed the amount of fully assembled complex I by immuno-detection on electroblotted blue-native polyacrylamide gels using an NDUFA9 antibody as described by Nijtmans et al.11
In patient 50845 carrying the NDUFB3 mutations, we additionally performed electrophoresis and in-gel quantification of fluorescent labelled proteins. Standard protocols for solubilisation and isolation of mitochondrial complexes12 13 were modified to improve quantification of mitochondrial complexes from human fibroblasts.14 Briefly, native proteins and protein complexes in homogenates from 10 mg fibroblasts (wet weight) were labelled with NHS-fluorescein (Thermo). Sediments containing mitochondrial membranes were resuspended in a buffer containing 50 mM NaCl, 50 mM imidazole, 2 mM aminohexanoic acid, and 1 mM EDTA, pH 7 and solubilised with 5 μl digitonin (20%). Following 1-D BNE (blue native electrophoresis) and 2D SDS-PAGE, the 2D gels were scanned using the blue laser (488 nm) of a Typhoon 9400 scanner (GE Healthcare) to detect fluorescein labelled proteins in mitochondrial complexes. 2D gels were further stained with silver.15 We used the DIA module (differential in-gel analysis) of the DeCyder 2D 7.0 software (GE Healthcare) for densitometric quantification of the fluorescence intensities in 2D gels. A few clearly visible signals of subunits from ATP synthase, complexes III and IV, the supercomplexes containing complex I, III, IV, and the porin complexes16 were selected for quantification (circled).
Results
Exome analysis
By combining unbiased exome analysis, sequential filtering, and functional investigation, we report here the identification of disease mutations in five genes in seven out of 10 index cases with isolated complex I deficiency. The first filter for known complex I deficiency disease alleles identified homozygous mutations in two individuals, 33463 and 59029. We found previously reported missense mutations in NDUFS317 (MIM 603864) and ACAD914 (MIM 611126), coding for a structural complex I subunit and a complex I assembly factor, respectively (table 3).
New genetic diagnoses for seven index cases with complex I deficiency
The second filter prioritised novel homozygous or compound heterozygous variants affecting genes encoding complex I subunits or known complex I assembly factors. Novel variants in such genes were identified in exomes from individuals 33284 (c.229C>T, p.Arg77Trp; c.476C>A, p.Ala159Asp in NDUFS8 (MIM 602141)), and 50845 (c.64T>C, p.Trp22Arg; c.208G>T, p.Gly70X in NDUFB3 (MIM 603839)) (table 3). Mutations in both genes have been previously shown to cause complex I deficiency.19–21 In individual 33027 we identified variants in two genes encoding complex I subunits, NDUFS7 (c.124C>T, p.Pro42Ser; c.514C>A, p.Arg172Ser (MIM 161825)) and NDUFS8 (homozygous c.187G>C/ p.Glu63Gln). Since the equally affected sister (44559) only carried the homozygous NDUFS8 mutation, we concluded that the NDUFS7 variants were not responsible for complex I deficiency.
DNA of available relatives was then tested. Concordant with a postulated pathogenic role of the NDUFS8 and NDUFB3 variants, affected individuals were homozygous (MITO021) or compound heterozygous mutants (MITO004, MITO005) (table 3), while the parents were heterozygous carriers.
In the remaining samples we applied a third filter for genes encoding mitochondrial proteins listed in MitoP2 (n=1239).8 This filter revealed that individual 44409 was compound heterozygous for a maternal mutation c.626C>T, p.Ser209Leu and a paternal mutation c.994C>T, p.Arg332X in MTFMT (MIM 611766), the gene encoding mitochondrial methionyl-tRNA-formyltransferase. MTFMT has so far not been associated with isolated complex I deficiency. However, the c.626C>T mutation was recently reported in a family with Leigh syndrome, impaired mitochondrial translation, and combined OXPHOS defects.18 The c.626C>T, p.Ser209Leu mutation was also identified in the homozygous state in patient 52075, with both reportedly non-consanguineous parents being heterozygous carriers. In addition to the predicted p.Ser209Leu change, c.626C>T has been demonstrated to cause skipping of exon 4 resulting in a frameshift and premature stop codon, p.Arg181SerfsX5.18 The second mutation found in individual 44409, c.994C>T, p.Arg332X, determines the synthesis of a truncated variant missing the highly conserved C-terminal portion of the protein. Thus, both mutations are shown or predicted to severely impair the function of MTFMT, supporting their pathogenic impact. In metazoan mitochondria, tRNAMET plays a role in both translationinitiation as well as translation elongation. MTFMT formylates Met-tRNAMet to form fMet-tRNAMet. Formylated methionine-tRNA has high affinity to, and is preferentially recruited by, mitochondrial translation initiation factor (IF2mt) to theribosomal P site for translation initiation.22
Confirmation of the pathogenicity of NDUFS8 and NDUFB3 mutations
The newly identified disease segregating missense mutations in NDUFS8 and NDUFB3 affected amino acid residues conserved in >85% of at least 39 vertebrates. Nevertheless, their pathogenic role remained to be proven. To this aim, complementation experiments were performed followed by functional and protein level investigations. Expression of NDUFS8wt cDNA in fibroblasts of individuals 33284 and 33027, and NDUFB3wt in cells of patient 50845, led to a significant increase of complex I activity (figure 2). In the same samples, we also analysed the amount of fully assembled complex I by blue-native PAGE electrophoresis. The complex I content was consistently low in naïve cells, but increased significantly after expression of the corresponding wild type cDNA (figure 2).

Introduction of NDUFS8 and NDUFB3 cDNA rescues both amount and activity of complex I in subject fibroblasts. The mutations in NDUFS8 and NDUFB3 result in decreased activity and amount of complex I. (A) 30 μg digitonin-solubilised protein from control and patients 33027, 33284, 50845 fibroblasts were separated on a 4–16% acrylamide blue-native PAGE (Novex System, Invitrogen), transferred on PVDF membrane and immuno-decorated with antibodies specific for subunits NDUFA9 of complex I and core II of complex III (1:1000; MitoSciences, Eugene, Oregon, USA). The analysis shows reduced amounts of subunit NDUFA9 assembled in supercomplex 0 and 1 (S0, S1) in patient cell lines indicating a decreased amount of whole complex I. (B) Control and patient fibroblasts were seeded at 20 000 cells/well in 80 μl DMEM, and incubated at 37°C, 5% CO2 for 24 h. Oxygen consumption rates (OCR) were measured using the XF96 extracellular flux analyser (Seahorse bioscience). The OCR was determined with no additions, and after addition of oligomycin (1 μM), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, 0,4 μM), and rotenone (5 μM). Complex I activity was calculated as rotenone-sensitive OCR. Both amount and activity of complex I are rescued upon lentiviral-mediated stable expression of NDUFS8wt and NDUFB3wt cDNA. Data shown are mean of >10 biological replicates ± SD. ***p<0.001.
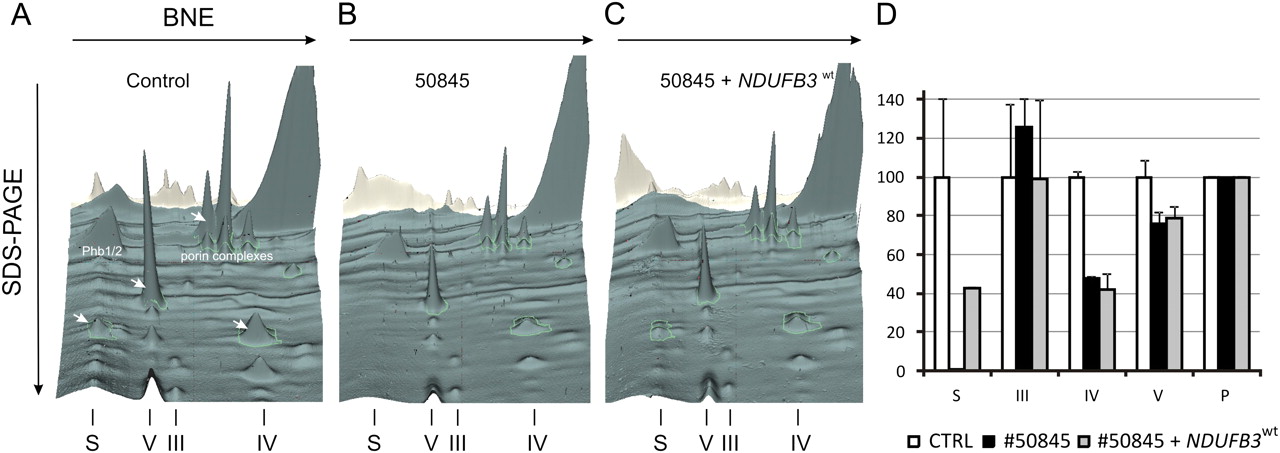
Since expression of the NDUFB3wt cDNA re-established complex I dependent respiration to low normal levels, we further tested the functional consequence of the identified variants in NDUFB3. The low complex I activity found in naïve cells of patient 50845 (17% of controls) remained unchanged after expression of NDUFB3 cDNA carrying either the p.Trp22Arg or the p.Gly70X mutations (figure 3). In parallel we also showed that the NDUFB3 protein was absent in naïve cells of patient 50845, but present in an amount comparable to controls after transduction with the NDUFB3wt cDNA. The same effect was also observed for the NDUFB8 and NDUFS3 subunits, indicating a general assembly/stability defect of complex I. The impaired complex I assembly or stability due to NDUFB3 mutations was further confirmed by two-dimensional blue-native/SDS-PAGE separation and quantification of fluorescein-labelled mitochondrial complexes (figure 4). Comparing patient and control fibroblast, the signal intensity of complex III was of the same level. Complex IV and V of patient fibroblasts were reduced to 48% and 76%, respectively. The supercomplexes were not detectable in the patient. After the transduction of the patient cell line with the NDUFB3wt, the amounts of mitochondrial supercomplexes in patient fibroblasts increased up to 43% of the control. The amounts of complexes III, IV, and V did not change significantly.
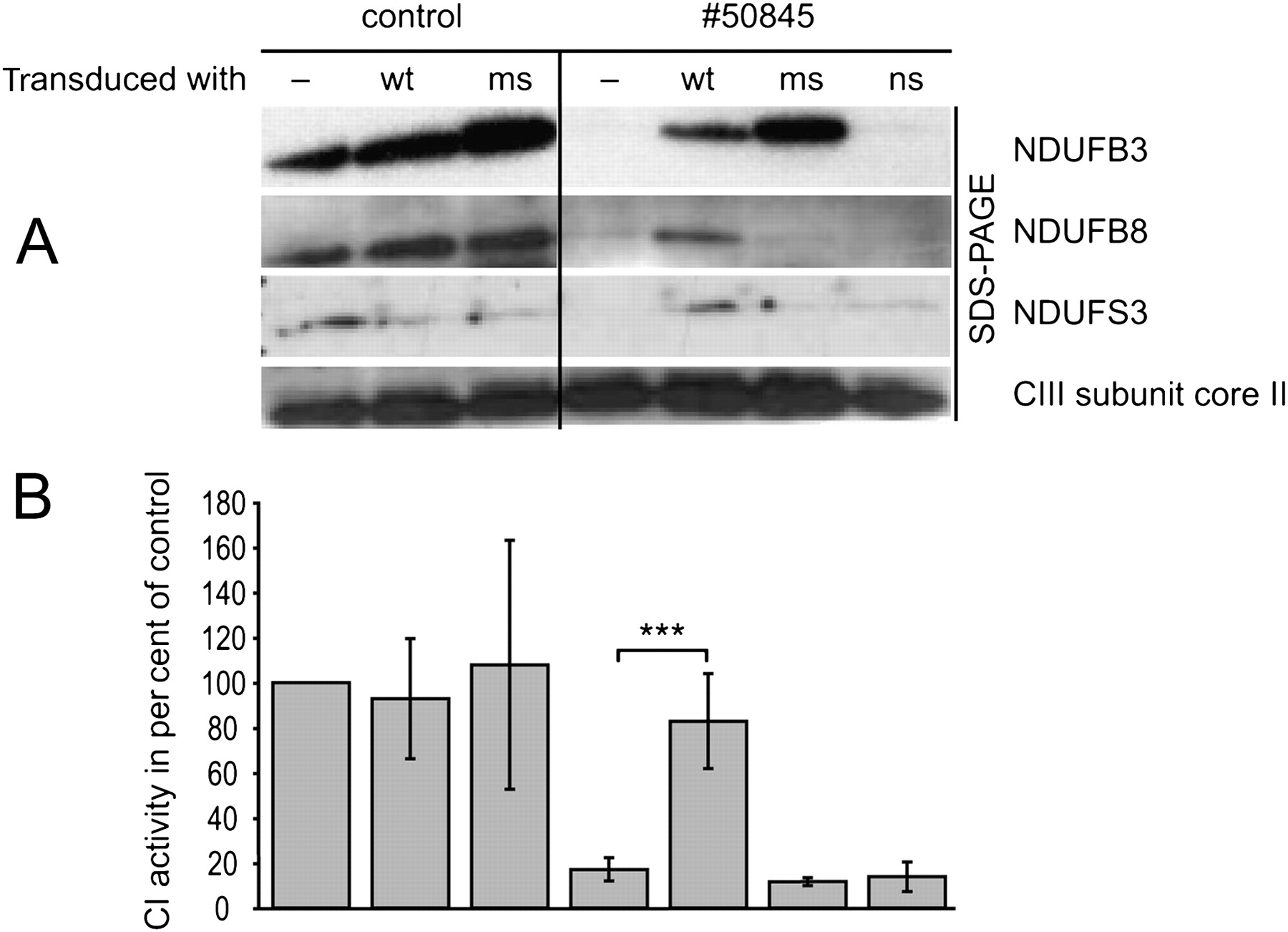
Expression of mitochondrial complex I subunits/complex I assembly. For Western-blot analysis 20 μg of total cell protein from patient 50845 and control (Ct) skin cultured fibroblasts with ectopic expression of wild type (wt) form or mutated versions of NDUFB3 cDNA were separated and immuno-decorated with antibodies specific for subunits of the respiratory chain complex I (NDUFB3, NDUFB8, NDUFS3) and subunit core II of complex III as control (MitoSciences 1:1000). Only expression of NDUFB3wt cDNA rescued complex I (A) and restored normal levels of several other complex I subunits (B). ms, NDUFB3 cDNA carrying the c.64T>C missense variant; ns, NDUFB3 cDNA with the c.208G>T stop mutation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mito-Panorama: two dimensional (2D) BN/SDS-PAGE separation and quantification of fluorescein labelled mitochondrial complexes and supercomplexes. For analysis we used 10 mg of control fibroblasts (A) and patient 50845 fibroblasts before (B) and after (C) transduction with the NDUFB3wt construct. 2D gel area for 3D visualisation see supplementary figure 1. Assignment of complexes: S, supercomplexes composed of respiratory chain complexes I, III, and IV, V, complex V or ATP synthase; III, complex III or cytochrome c reductase; IV, complex IV or cytochrome c oxidase; Phb1/2, prohibitin complex and P for porin complexes, which were used for normalisation. D, densitometric quantification of mitochondrial complexes in 2D BN/SDS-PAGE from human fibroblasts, expressed as per cent of healthy control cells (two technical replicates). Arrows indicate fluorescent signals used for complex quantification that are visible in this 3D view.
Taken together, these results demonstrate the causal role of the mutations identified in NDUFS8 and NDUFB3, thereby establishing NDUFB3 as a gene responsible for human complex I deficiency.
Discussion
In this study, increasingly wider variant filters were applied on exome sequencing datasets, in combination with functional complementation, to identify causative mutations in single individuals with complex I deficiency. Filtering of rare variants was facilitated by the availability of a large set of control exomes (n=879), and pathogenic mutations were identified using mutation databases and in silico selection of genes encoding known or potential mitochondrial proteins. Importantly, when the list of genes was further processed against public databases—for example, exclusion of variants present in dbSNP with an average heterozygosity of more than 0.02—the number of candidate genes was reduced by just one or two entries. The only exception was a sub-Saharian African individual (33027), whose gene variation dataset matched poorly with our set that was largely composed of Europeans.
Our stepwise filtering approach successfully identified variants in NDUFB3. Complementation experiments established the pathogenicity of the mutations. Two previous large candidate-gene based screening studies failed to identify rare DNA variants in the NDUFB3 gene among 1035 and 1526 patients with complex I deficiency. NDUFB3 variants were found only recently in a screen of 42 patients19 with infantile onset mitochondrial disorders, suggesting that mutations in NDUFB3 are rare. NDUFB3 is a single transmembrane domain subunit localised in the QD module of the complex I membrane arm—being one of the many complex I accessory subunits23 present in eukaryotes but not in bacterial complex I—which play a role in regulation, assembly and stability of the 14 subunit catalytic core of complex I, possibly protecting it from reactive oxygen species damage.24 The ancillary but essential role of the complex I accessory subunits is confirmed by the identification of mutations resulting in impaired complex I activity in eight of them, including NDUFB3 (present study).
Filtering for genes encoding mitochondrial proteins revealed loss-of-function mutations in MTFMT in two patients. The very same disease allele, c.626C>T, shared by patients 44409 and 61606 has also been reported in a patient presenting with a biochemical signature of combined respiratory chain deficiency.18 MTFMT is a protein involved in mtDNA translation, which suggests that complex I deficiency can be a biochemical signature common to mutations of genes neither directly nor exclusively involved in complex I formation and activity. Giant size, structural complexity and intricate functional and structural interactions with other respiratory chain components and mitochondrial pathways are likely concurring to make complex I an exquisitely sensitive target in a wide spectrum of pathological conditions related to OXPHOS. For example, seven of the 13 mtDNA encoded proteins are complex I subunits, which could explain why mtDNA translation defects such as those associated with mutations in MTFMT can predominantly impair complex I activity (table 4).
Biochemical analysis of OXPHOS complexes in muscle biopsy in individuals 44409 and 61606 harbouring MTFMT mutations
These considerations have relevant implications for the diagnostic workup and for the elucidation of complex I functions and interactions.
Three individuals remained genetically undefined even after exome analysis, which may be due to: (1) insufficient gene coverage; (2) too stringent filtering; (3) mutations in non-coding regions or non-annotated exons; (4) heterozygous deletions of whole exons; or (5) non-autosomal recessive mode of inheritance. However, the third filter detected between 0–5 mitochondrial proteins. We are currently analysing the candidate genes in the unsolved cases. An interesting finding of our present study confirming a previous report14 is the identification of unexpected disease genes associated with complex I pathophysiology: the first example has been the recently identified gene encoding ACAD9, a bona fide component of the β oxidation of fatty acids, followed here by MTFMT. Along the same line, the individuals that escaped the mitochondrial filter are of special interest, since they offer the possibility to discover mutations in genes so far unlinked to mitochondrial function. This option, important for many patients, would have been missed in an approach focused on genes predicted to code for mitochondrial proteins only.19
In summary, this study demonstrates the efficacy of exome sequencing, coupled with prioritisation for mitochondrial proteins and experimental validation, to elucidate the genetic basis of a metabolic disorder with extreme locus heterogeneity. This integrated strategy can be further exploited to define fully the gene repertoire involved in complex I deficiency and in other mitochondrial disorders displaying a biochemical phenotype at the cellular level. In some cases, as for family MITO053 with mutations in ACAD9, the identification of the disease gene offers the pathophysiological basis for promising therapeutic interventions—for example, the high dose supplementation with riboflavin.14 25 26
Web resources
Acknowledgments
We thank the patients and their families for their participation in the study, and R Hellinger, E Botz, and C Fischer for technical support.
References
Supplementary materials
HTML Page - index.htslp
Footnotes
Funding This work was supported by the Impulse and Networking Fund of the Helmholtz Association in the framework of the Helmholtz Alliance for Mental Health in an Ageing Society (HA-215), the German Federal Ministry of Education and Research (BMBF) funded Systems Biology of Metabotypes grant (SysMBo #0315494A), and the German Network for Mitochondrial Disorders (mitoNET #01GM0867 and 01GM0862). TM is supported by the BMBF funded German Center for Heart Research. TM and TMS were supported by the European Commission 7th Framework Program, Project N. 261123, GEUVADIS and the German Ministry for Education and Research (01GR0804-4). JAM was supported by the Wissenschaftspreis 2008 of the Austrian Paediatric Society (ÖGKJ), WS was supported by the Jubiläumsfonds of Oesterreichische Nationalbank (#12568) and JAM, FZ and WS were supported by the Vereinigung zur pädiatrischen Forschung und Fortbildung Salzburg. MT, HH, and JZ were supported by research project PRVOUK of the Charles University in Prague - First Faculty of Medicine (program MOLMED. VS was supported by the Bundesministerium für Bildung und Forschung (BMBF 01GM0863; mitoNET) and IW by the Deutsche Forschungsgemeinschaft, Sonderforschungsbereich 815, Project Z1 (Redox-Proteomics). MZ was supported by Telethon GPP 10005, Telethon GGP11011, Cariplo 2011-05-26.
Competing interests None.
Patient consent Obtained.
Ethics approval Ethics approval was provided by the ethics committee of the medical faculty of the Technical University of Munich. Written informed consent was obtained from all study participants or their guardians and the study was approved at the recruiting centre.
Provenance and peer review Not commissioned; externally peer reviewed.