Article Text

Abstract
Background The genetic causes for most male infertility due to severe asthenozoospermia remain unclear.
Objective Our objective was to identify unknown genetic factors in 47 patients with severe asthenozoospermia from 45 unrelated Chinese families.
Methods We performed whole exome sequencing of 47 individuals with severe asthenozoospermia from 45 unrelated families. Mutation screening was performed in a control cohort of 637 individuals, including 219 with oligoasthenospermia, 195 with non-obstructive azoospermia and 223 fertile controls. Ultrastructural and immunostaining analyses of patients’ spermatozoa were performed to characterise the effect of variants.
Results One homozygous non-sense mutation (NM_194302, c.G5341T:p.E1781X), two compound heterozygous mutations (c.C2284T:p.R762X and c.1751delC:p.P584fs) and two compound heterozygous mutations (c.5714_5721del:p.L1905fs and c.C3021A:p.N1007K) were identified in CFAP65 of three individuals with completely immotile spermatozoa, respectively. No biallelic deleterious variants of CFAP65 were detected in the control cohort of 637 individuals. Ultrastructural and immunostaining analyses of spermatozoa from two patients showed highly aberrant sperm morphology with severe defects such as acrosome hypoplasia, disruption of the mitochondrial sheath and absence of the central pair complex.
Conclusion To the best of our knowledge, we are the first to report that CFAP65 mutations may cause spermatozoa to be completely immotile.
- male infertility
- completely immotile spermatozoa
- CFAP65 mutation
- acrosome
- flagellum assembly
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Infertility affects over 186 million people worldwide, with men and women being impacted equally.1 Male infertility is often caused by asthenozoospermia, which is characterised by highly reduced sperm motility or sperm immobility. Two common forms of asthenozoospermia which can be traced back to a heterogeneous genetic origin have been identified as follows: primary ciliary dyskinesia (PCD), a multisystem disorder (eg, chronic rhinosinusitis, bronchiectasis or heterotaxis) caused by dysfunction of motile cilium and flagellum2; and multiple morphological abnormalities of the sperm flagella (MMAF), a characteristic of ejaculated spermatozoa with mosaic morphological abnormalities of the flagella.3 Asthenozoospermia cases constitute a heterogeneous group, where approximately 20 PCD-related and 11 MMAF-related asthenozoospermia genes have been identified.4 5 However, the genetic aetiology of these conditions remains unclear.
Complete sperm immobility is the most severe form of asthenozoospermia, as affected couples are only able to conceive via intracytoplasmic sperm injection (ICSI). Furthermore, the potential for transmission of this disease to offspring is a leading concern.6 Therefore, the identification of genetic causes combined with ultrastructural evaluation is not only pivotal for revealing mechanisms underlying spermiogenesis in these patients but also vital for improving treatment choices and providing adequate genetic counselling for patients.
Although acrosomal anomaly is not frequently described in relation to asthenozoospermia, it is strongly associated with male infertility, since lower fertility, pregnancy and live birth rates associated with ICSI have also been reported in these cases.7 Acrosomal defects are categorised as acrosomal hypoplasia and acrosomal absence (also known as globozoospermia).8 Its overall prevalence is currently unknown as acrosomal hypoplasia is largely underdiagnosed and frequently remains clinically unidentified,9 although the incidence of globozoospermia appears to be comparatively low (0.1%). Moreover, despite the availability of a large list of mouse candidate genes involved in acrosome defects, mutations in only three human orthologues (PICK1, SPATA16 and DPY19L2) have been reported and all were associated with globozoospermia.10 11 ,12
The current study reports findings in 47 individuals with severe asthenozoospermia from 45 unrelated families, where biallelic CFAP65 mutations were identified in three individuals who presented sperm flagellar defects and acrosomal hypoplasia.
Materials and methods
Study subjects
All subjects in this study were recruited from the Reproductive and Genetic Hospital of CITIC-Xiangya from 2014 to 2018. Following a detailed evaluation of medical history, 47 affected males (from 45 families) were selected from among 1752 infertile males who presented with >95% immotile sperms (the percentage of progressively motile sperm <5%), including 44 patients with completely immotile spermatozoa and 3 patients with 95%–100% immotile sperms. Nine individuals were from consanguineous families. A random cohort of 637 individuals of matching ages, including 219 with oligoasthenospermia (sperm counts <2×106/mL, progressively motile sperm <32%), 195 with non-obstructive azoospermia and 223 controls with normal fertility (have at least one offspring by natural fertilisation), were recruited in this study for mutation screening. Abnormalities in somatic karyotypes and azoospermia factor microdeletion on the Y chromosome were not detected. Other causes of infertility, such as reproductive malformation, drugs and exposure to gonadotoxic factors, were also excluded. Three independent semen evaluations were performed according to WHO (2010) guidelines.13
The present study was approved by the Institutional Ethics Committees of Central South University and the Reproductive Genetic Hospital of CITIC-Xiangya, China. All methods were performed in accordance with approved guidelines. Written informed consent was obtained from all individuals who participated in the study prior to commencement.
Whole exome sequencing (WES) and bioinformatic analysis
Blood samples from participating individuals, including those of 47 individuals with severe asthenozoospermia and 637 control individuals, were collected and genomic DNA was extracted using a DNA extraction kit (Qiagen, Hilden, Germany). This genomic DNA was subsequently used for WES analysis. Library construction, WES and data analysis were carried out by the Beijing Genome Institute at Shenzhen as described previously.14 Variant identification was performed using the Genome Analysis Toolkit package. Sequencing variants, including single-nucleotide variants and small insertions or deletions, were annotated using ANNOVAR software.15
The most promising candidate pathogenic variants in 47 affected individuals were screened using the following criteria: variants with a minor allele frequency below 5% in the public databases (dbSNP, 1000 Genomes Project and ExAC); predicted to be deleterious using different tools (Polyphen-2 (genetics.bwh.harvard.edu/pph2), MutationTaster (www.mutationtaster.org/), SIFT (sift.jcvi.org/) and Combined Annotation Dependent Depletion (cadd.gs.washington.edu/)); phenotype relevance using comprehensive expression data (high expression in testis) and model organism data.16 Meanwhile, for patient from consanguineous family, we performed homozygosity mapping using the Homozygosity Mapper,17 and the homozygous variants located in >5.0 Mb homozygous regions were considered in priority. WES data from the 637 controls were used to screen candidate gene mutations.
Sanger sequencing
Specific PCR primers were designed to the region of the mutations in the candidate gene CFAP65 by using the following sequences: CFAP65-F1: 5′-CAATCCTGCCTGTACCCTCC-3′, CFAP65-R1: 5′-AACCTTCTGCACTTCGCCTC-3′; CFAP65-F2: 5′-GCAGTTGAGCCCTATCCACC-3′, CFAP65-R2: 5′-CACCGTGTAAAGGCAGTTGG-3′; CFAP65-F3: 5′-GACACAGGACTCTGAGCTTTTC-3′, CFAP65-R3: 5′-CTGTTGGGGCCACGCTTAC-3′; CFAP65-F4: 5′-TTCTGCGTGCCCAGGTAAGG-3′, CFAP65-R4: 5′-CTAGCGGCATGTCGGAGAG-3′; CFAP65-F5: 5′-CCACCACCCACTACATGCTC-3′, CFAP65-R5: 5′-TAACCGGTCTTAACCAGTACCT-3′. Amplified PCR products were analysed via 2.0% agarose gel electrophoresis to determine band size, following which bidirectional sequencing was performed using an ABI 3730 automated sequencer (Applied Biosystems, Foster City, California, USA).
Expression analysis
Total RNA was extracted from different tissues (brain, lung, heart, spleen, kidney, stomach, colon, testis, ovary, skin, muscle) of wild-type C57BL/6 adult mice using TRIzol reagent (Invitrogen, Carlsbad, California, USA), and 1 µg RNA was reverse transcribed into cDNA using a goScript reverse transcription system (Promega, Madison, Wisconsin, USA) according to the manufacturer’s instructions. Evaluation of mouse Cfap65 gene expression was performed via PCR amplification with 1 µL cDNA. The mRNA expression levels of Cfap65 were normalised to those of the Actb using the following primers: Cfap65, F:5′-GCATAACGCTCACACTTCCC-3′, R:5′-CTGAAATGGGGCTGGGACTT-3′ and Actb, 5′-AGATCAAGATCATTGCTCCTCC-3′ and 5′-AGCTCAGTAACAGTCCGCCT-3′. PCR conditions were as follows: 95°C for 10 min; 35 cycles of 95°C for 30 s, 57°C for 30 s and 72°C for 30 s; and 72°C for 10 min. PCR products were validated via Sanger sequencing. The assays were repeated thrice.
Scanning electron microscopy (SEM)
Semen samples from normal controls and the patients (F1: II-2 and F2: II-2) were prepared as previously described.18 Spermatozoa were fixed in 2.5% phosphate-buffered glutaraldehyde at 4°C for 2 hours. Immobilised spermatozoa were deposited on poly-L-lysine coated coverslips. The coverslips were washed in distilled water, dehydrated via an ascending gradient of cold 50%, 70%, 95% and 100% ethanol, and dried at critical point using a Quorum K850 Critical Point Dryer (East Sussex, UK). Specimens were then attached to specimen holders and coated with gold particles using an ion sputter coater (Q150RS Rotary-Pumped, Quorum Technologies, East Sussex, UK) before being viewed with an S-3400N scanning electron microscope (Hitachi, Tokyo, Japan).
Transmission electron microscopy (TEM)
Semen samples of normal controls and patients (F1: II-2 and F2: II-2) were treated as previously described.14 Samples were fixed with glutaraldehyde (Sigma-Aldrich, St. Louis, Missouri, USA) and osmium tetroxide, followed by OsO4 and sucrose, and dehydrated using graded ethanol. Subsequently, the samples were embedded in Epon 812, dodecenylsuccinic anhydride, methylnadic anhydride and dimethylaminomethyl phenol. Ultrathin (70–90 nm) sections were contrasted with uranyl acetate and lead citrate. An HT7700 Hitachi electron microscope and a MegaView III digital camera (Munster, Germany) were used for image capturing.
Histological analysis
For H&E staining, semen smears were dehydrated with graded ethanol, stained with H&E, dehydrated again with graded ethanol and processed with dimethylbenzene for 5 min twice. For histological analysis, human testicular tissue from a patient with prostate cancer with normal fertility were fixed in Bouin’s solution (Sigma-Aldrich), embedded in paraffin, sectioned, processed, saved and used for subsequent experiments. Immunofluorescence analysis was performed as previously described.19 The slides were incubated with primary antibodies (CFAP65, SPAG6 and anti-acetylated tubulin monoclonal antibody) for 2.5 hours at 37°C. The details of all antibodies are listed in online supplementary table S1. The slides were then incubated with secondary antibodies (Alexa Fluor 488 anti-mouse IgG (A-21121, 1:400) and Alexa Fluor 555 anti-rabbit IgG (A31572, 1:400)) for 1.5 hours at 37°C. For evidence of the acrosome, the samples were treated with Pisum sativum agglutinin (PSA) and incubated for 2.5 hours at 37°C. Finally, all slides were stained using 2-(4-amidinophenyl)-1H-indole-6-carboxamidine for 5 min at room temperature. An Olympus IX51 fluorescence microscope (Olympus, Tokyo, Japan) and VideoTesT-FISH 2.0 software (VideoTesT, Petersburg, Russia) were used for photographing fluorescence signals.
Supplemental material
ICSI procedures
The wives of three individuals with CFAP65 mutations (F1: II-2, F2: II-2 and F3: II-1) accepted ICSI treatment. They underwent controlled ovarian hyperstimulation, oocyte retrieval, ICSI and pregnancy confirmation as described previously.20 21 Viable spermatozoa for ICSI, selected from completely immotile sperm, were stimulated using the SperMagic medium.20 21 Pregnancy was confirmed via two human chorionic gonadotropin tests conducted 14 day following embryo transfer, where clinical pregnancy was defined as at least a visible sac with a fetal heart beat detected via ultrasound screening after 28 days.
Results
CFAP65 is a candidate gene for male infertility with completely immotile spermatozoa in humans
In the present study, WES was performed to analyse the genetic aetiology of a cohort of 47 individuals with severe asthenozoospermia from 45 unrelated Chinese families. After exclusion of frequent variants and applying filtering criteria described in the Materials and methods section (online supplementary table S2), we identified three individuals with biallelic variants in CFAP65 (figure 1A), accounting for 6.7% (3/45) of our cohort (online supplementary table S3). The first proband (F1: II-2, 36 years old) from a consanguineous family (family 1) presented with severe asthenoteratospermia with 100% immotile spermatozoa and a comparatively low sperm count (14.5–15.3×106/mL) (online supplementary table S4). F1: II-2 exhibited classic PCD-like symptoms from early life, including a positive history of chronic wet cough, nasal obstruction and pneumonia, and when these individuals reached adulthood, these symptoms gradually developed into recurrent airway infections, bronchitis and rhinosinusitis (online supplementary figure S1A). However, F1: II-2 refused to perform a ciliary biopsy or a nasal nitric oxide test. Routine semen analysis of F2: II-2 (26 years old) from a non-consanguineous family (family 2) also revealed severe asthenoteratospermia with 100% immotile spermatozoa and a comparatively low sperm count (11.3–13.4×106/mL) (online supplementary table S4). F2: II-2 also exhibited a positive history of chronic wet cough, nasal obstruction and pneumonia. However, paranasal sinus and chest CT scans of F2: II-2 were normal without any abnormalities (online supplementary figure S1B). The third affected individual F3: II-1 (39 years old) was also from a non-consanguineous family and diagnosed with oligoasthenoteratospermia (figure 1A). Semen analysis showed a reduced sperm count (5.3–6.9×106/mL) with 100% sperm immobility (online supplementary table S4). No respiratory symptoms were found in F3: II-1. Bilateral testicular size, measured via B ultrasonography, was normal and no abnormal hormone levels were detected (online supplementary table S4).
Supplemental material

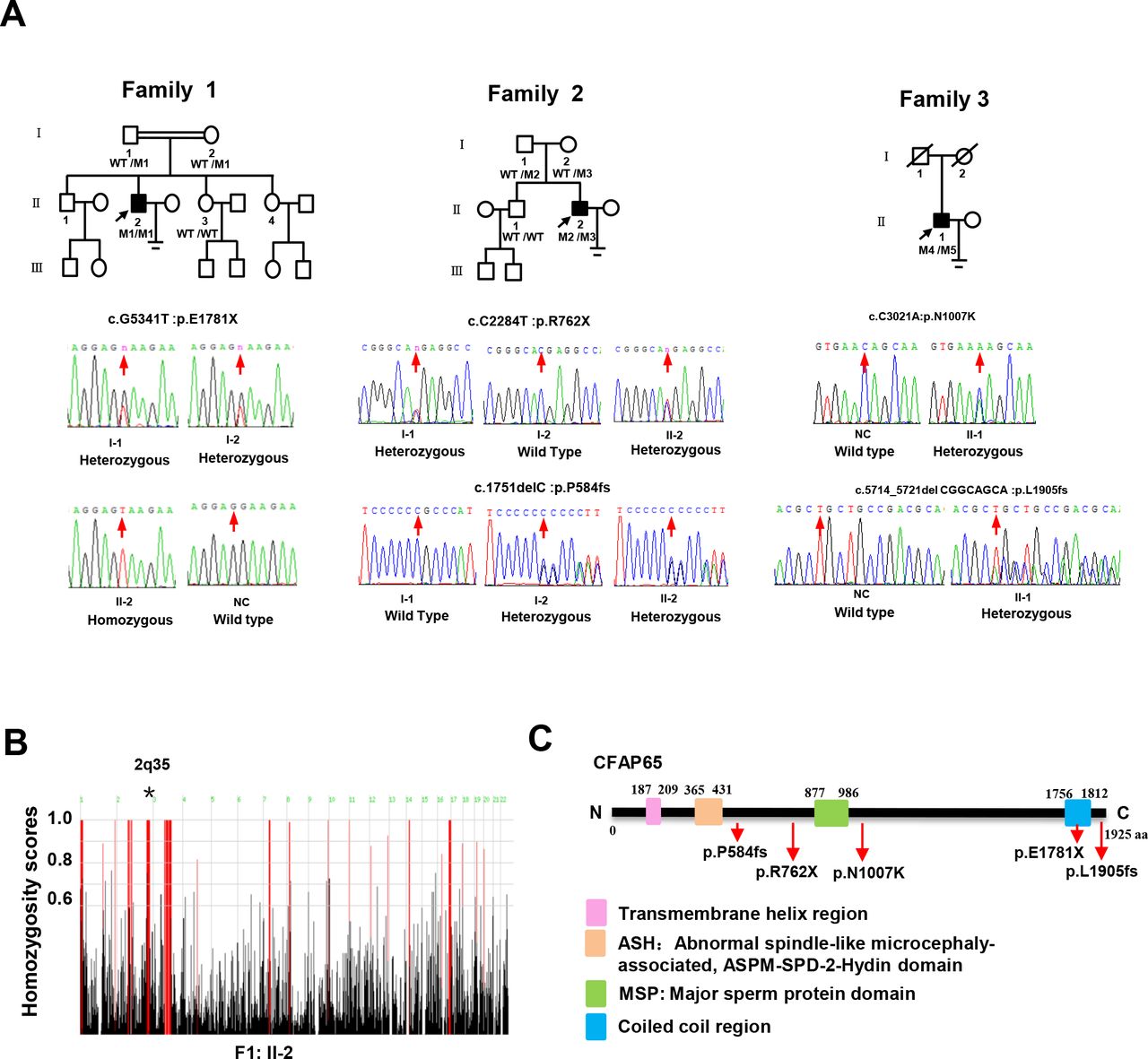
The genotypic features of the affected individuals. (A) The pedigrees of three families with inherited CFAP65 mutations. Probands in family 1, F1: II-2; family 2, F2: II-2 and family 3, F3: II-1 are marked with a black arrow. CFAP65 genotype of each individual is indicated with WT indicating a normal allele and M1/M2/M3/M4/M5 indicating mutant alleles. (B) Homozygosity mapping of F1: II-2. Red colour indicates the most promising homozygous regions. An asterisk (*) indicates the area where CFAP65 is located. (C) Location and nature of CFAP65 mutations in CFAP65 protein. Pink square represents the transmembrane helix region; orange square represents the ASPM-SPD-2-Hydin (ASH) domain; green square represents the major sperm protein (MSP) domain and blue square represents the coiled-coil region. NC, normal control; WT, wild type.
The first homozygous non-sense variant of CFAP65 (GenBank: NM_194302, c.G5341T: p.E1781X) was confirmed in F1: II-2 from a consanguineous family via Sanger sequencing, whereas the unaffected parents (F1: I-1 and F1: I-2) were heterozygous carriers of this same CFAP65 variant (figure 1A), which was consistent with genotype–phenotype cosegregation. The genotypes of the fertile sisters were not obtained due to the unavailability of DNA samples. The second group of compound heterozygous variants in CFAP65 (c.C2284T: p.R762X; c.1751delC: p.P584fs) were detected in F2: II-2 from a non-consanguineous family by Sanger sequencing (figure 1A), in which one (c.C2284T: p.R762X) was a non-sense variant and the other was a frameshift variant (c.1751delC: p.P584fs). His father (F2: I-1) carried one heterozygous variant (c.C2284T), while his mother (F2: I-2) carried the other (c.1751delC). Their unaffected brother (F2: II-1) harboured the wild-type allele. The mode of inheritance was consistent with an autosomal recessive mode of inheritance. The third compound heterozygous variants (c.5714_5721del:p.L1905fs and c.C3021A:p.N1007K) in F3: II-1 were also confirmed via Sanger sequencing. Unfortunately, cosegregation analysis was not performed as blood samples of this individual’s parents could not be obtained. All identified CFAP65 variants were either absent or very rare in public databases (online supplementary table S3), and no deleterious biallelic variants in CFAP65 were detected in the control cohort of 637 individuals. Moreover, homozygosity analysis of patient F1: II-2 revealed that the CFAP65 mutation was located in the absence of heterozygous regions with a size greater than 5 Mb (figure 1B). Therefore, we speculated that novel homozygous and compound heterozygous variants of CFAP65 were the likely causes of this malady.
Morphological abnormalities were observed in sperm flagella and heads in CFAP65 mutant individuals
We investigated morphological and ultrastructural changes in the spermatozoa of CFAP65 mutant individuals. As the semen sample of F3: II-1 was not available, we were only able to perform subsequent phenotypic analyses of spermatozoa from F1: II-2 and F2: II-2. Under light microscopy, most spermatozoa of the affected individuals, F1: II-2 and F2: II-2, exhibited multiple tail abnormalities, including absent, short, bent, coiled or looped tails, compared with those of healthy control spermatozoa (figure 2A). When observed by SEM, spermatozoa of normal controls exhibited a smooth, regularly contoured, oval-shaped head and a long flagellum with a clearly defined midpiece, principal piece and end piece (figure 2B), whereas all sperm flagella of CFAP65 mutant individuals, F1: II-2 and F2: II-2, exhibited severe morphological defects. Of the CFAP65 mutant spermatozoa, 72.3% and 68.6% had misshapen heads (a total of 166 and 172 spermatozoa were observed, respectively). In general, head morphology defects ranging from mild to severe, such as an amorphous, tapered or small head, were observed in both individuals (figure 2B,I–VI), indicating a defect in sperm head shaping or acrosome formation. Sperm tails displayed unidentifiable or swollen midpieces and shorter principal pieces of highly irregular width, indicating poorly assembled mitochondrial sheaths (MS) and misarranged fibrous sheaths (FS). Flagella were frequently missing, and cytoplasmic residuals were occasionally observed in coil-shaped flagella (figure 2B,I–VI).
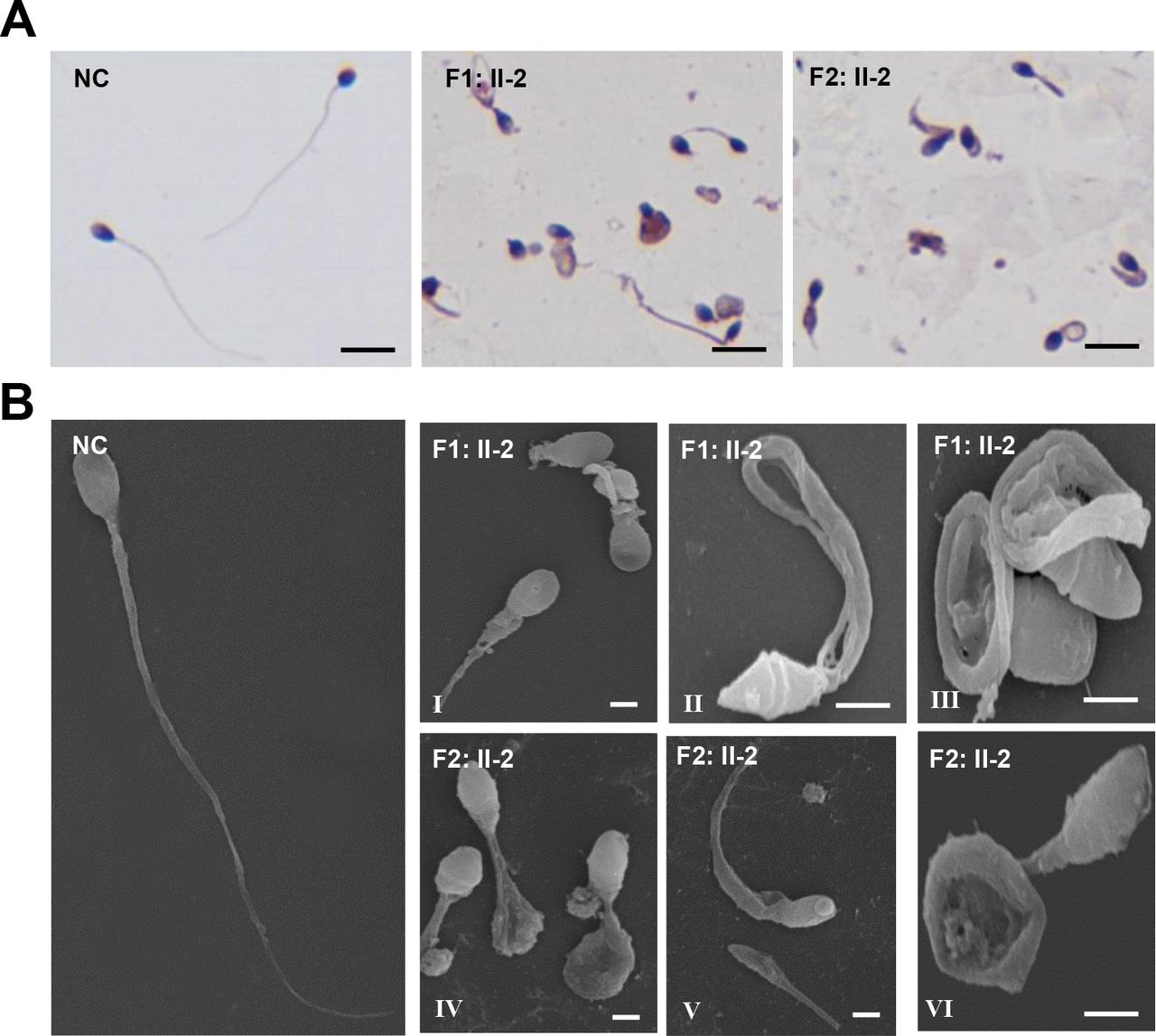
The morphology of CFAP65 mutant spermatozoa in two affected individuals. (A) H&E staining analysis of the morphology of spermatozoa from NC and CFAP65 mutant individuals (F1: II-2 and F2: II-2). All CFAP65 mutant spermatozoa have much shorter or irregular flagella than those of NC. Scale bars represent 10 µm. (B) Scanning electron microscopy of the spermatozoa of NC and CFAP65 mutant individuals (F1: II-2 and F2: II-2). Compared with spermatozoa with normal morphology in NC, malformed heads, such as amorphous, tapered or small heads, were often observed in F1: II-2 and F2: II-2 (I–VI). Most spermatozoa from both affected individuals exhibited flagella that are short, coiled, bent or had irregular width (I–VI). Scale bars represent 1 µm. NC, normal control.
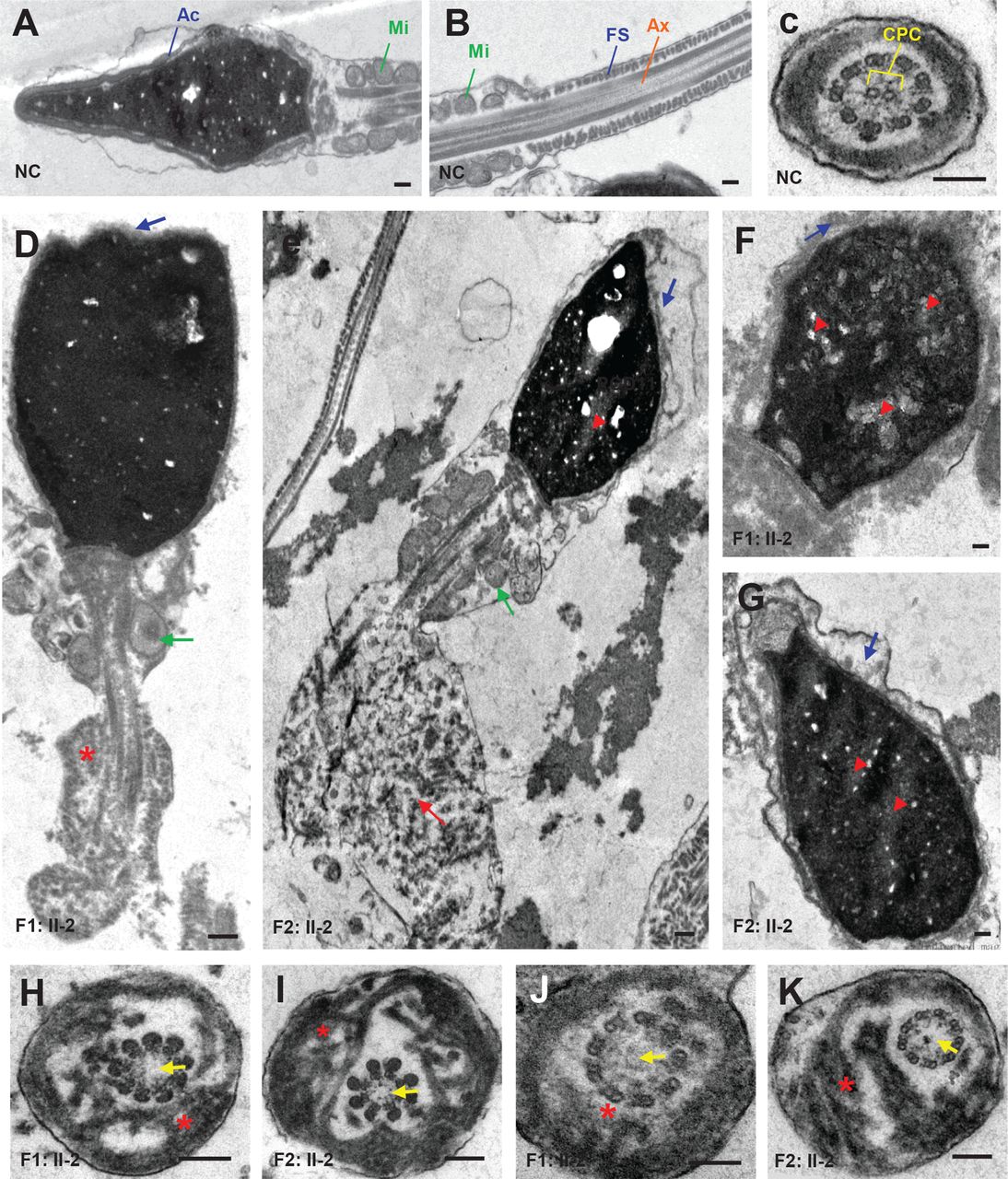
To better understand the nature of defects that were observed in sperm head and flagella of CFAP65 mutant individuals (F1: II-2 and F2: II-2), we analysed sperm ultrastructure using TEM (figure 3). Compared with normal controls (figure 3A–C), acrosomes of the spermatozoa of F1: II-2 and F2: II-2 were either absent or morphologically defective, while the sperm displayed abnormal head shapes (figure 3D–G). Most of the acrosomes were quite thin and showed highly diminished contents with barely recognisable inner and outer acrosomal membranes. Furthermore, a higher incidence of insufficient chromatin condensation was observed in the spermatozoa of affected individuals compared with those of normal controls. The most apparent defects observed in the longitudinal sections of flagella were poorly assembled or missing MS in the midpiece. Hyperplasia of FS was also observed (figure 3D–G). In cross sections (n≧20), 93.3% (28/30) of F1: II-2 and 83.9% (26/31) of F2: II-2 spermatozoa lacked a central pair complex (CPC) in the axoneme (9+0 structure) (figure 3F,G). The arrangement of FS was mostly disorganised. Overall, these results indicated that a lack of CFAP65 may lead to various defects in both the tail and head of spermatozoa.

Sperm ultrastructure in two CFAP65 mutant individuals. (A–C) The ultrastructure of longitudinal sections (scale bars=500 nm) and cross sections (scale bars=100 nm) of sperm flagella from NC. (D–G) The longitudinal sections (scale bars=500 nm) of spermatozoa from F1: II-2 and F2: II-2. Sperm tails with poorly assembled mitochondria (green arrow), disorganised FS (asterisk) or a cytoplasmic mass containing different components of the flagellum (red arrow) were observed. The acrosome was thin or broken with unidentifiable acrosomal membranes (blue arrow) along with misshapen heads; chromatin condensation seemed abnormal (red arrowhead). (H–K) Cross sections (scale bars=100 nm) of spermatozoa from F1: II-2 and F2: II-2. Spermatozoa lacked a CPC (yellow arrow) in the Ax. Disorganised FS were also frequently observed (asterisk). Ac, acrosome; Ax, axoneme; CPC, central pair complex; FS, fibrous sheath; Mi, mitochondria; NC, normal control.
CFAP65 is localised in the acrosome and flagellar midpiece
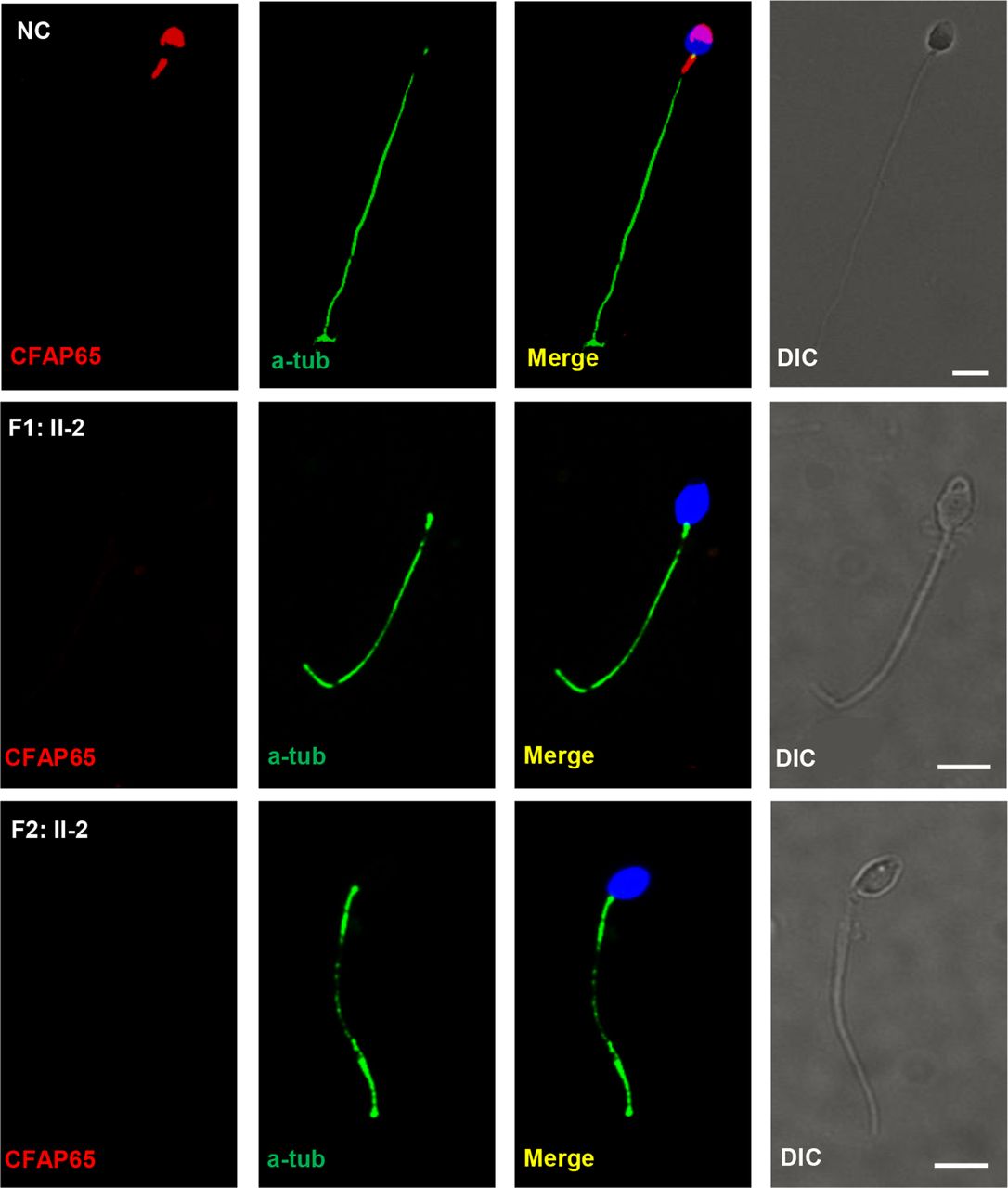
Human CFAP65 (also named CCDC108, MIM: 614270) is located on 2q35 and has 13 transcripts. The protein encoded by this gene is speculated to be a transmembrane protein, containing a transmembrane helix region near the N-terminus, an ASPM-SPD-2-Hydin (ASH) domain, a major sperm protein (MSP) domain and a coiled-coil region near the C-terminus (figure 1C). In this study, all three identified CFAP65 mutations in F1: II-2 and F2: II-2 were predicted to cause premature translation termination and produce truncated proteins (figure 1C). In such predicted CFAP65 truncated proteins, the coiled-coil region would be disrupted in F1: II-2, whereas both MSP domain and coiled-coil region would be eliminated in F2: II-2, indicating that the function of CFAP65 would be severely disrupted. Subsequently, we performed immunofluorescence analysis to compare CFAP65 expression in the spermatozoa of affected individuals, F1: II-2 and F2: II-2, with that of healthy controls. Spermatozoa from CFAP65-mutated individuals did not exhibit CFAP65 staining (figure 4). Importantly, CFAP65 staining was detected in the acrosome area and flagellar midpiece of normal spermatozoa (figure 4), which was consistent with TEM findings (figures 2 and 3). The above findings support a putative role for CFAP65 during acrosome formation and flagellum assembly. Immunostaining of human testes consistently showed that CFAP65 was specifically localised in the cytoplasm of round spermatid and elongating spermatid, but absent in the spermatocytes and Sertoli cells in the human testes (online supplementary figure S2A-B).

CFAP65 expression was absent in CFAP65 mutant spermatozoa. CFAP65 staining is present in the acrosome and flagellar midpiece of spermatozoa from normal controls (NCs), but absent in spermatozoa from F1: II-2 and F2: II-2. Scale bars represent 5 µm. DIC, differential interference contrast.
As phylogenetic evolutionary analysis showed a high homology between sequences of human and mouse CFAP65 (71%; https://blast.ncbi.nlm.nih.gov), we used mouse tissues to investigate the spatiotemporal expression pattern of Cfap65. The expression of Cfap65 in testes was particularly high from postnatal day 21 (online supplementary figure S3A) when haploid round spermatid began to morph into elongated spermatid, highlighting the crucial role of CFAP65 in spermiogenesis. Interestingly, besides being expressed in the testes, Cfap65 was also expressed in certain ciliated organs, such as the brain, lung and kidney (online supplementary figure S3B).
CFAP65 is essential for acrosome formation and flagellum assembly
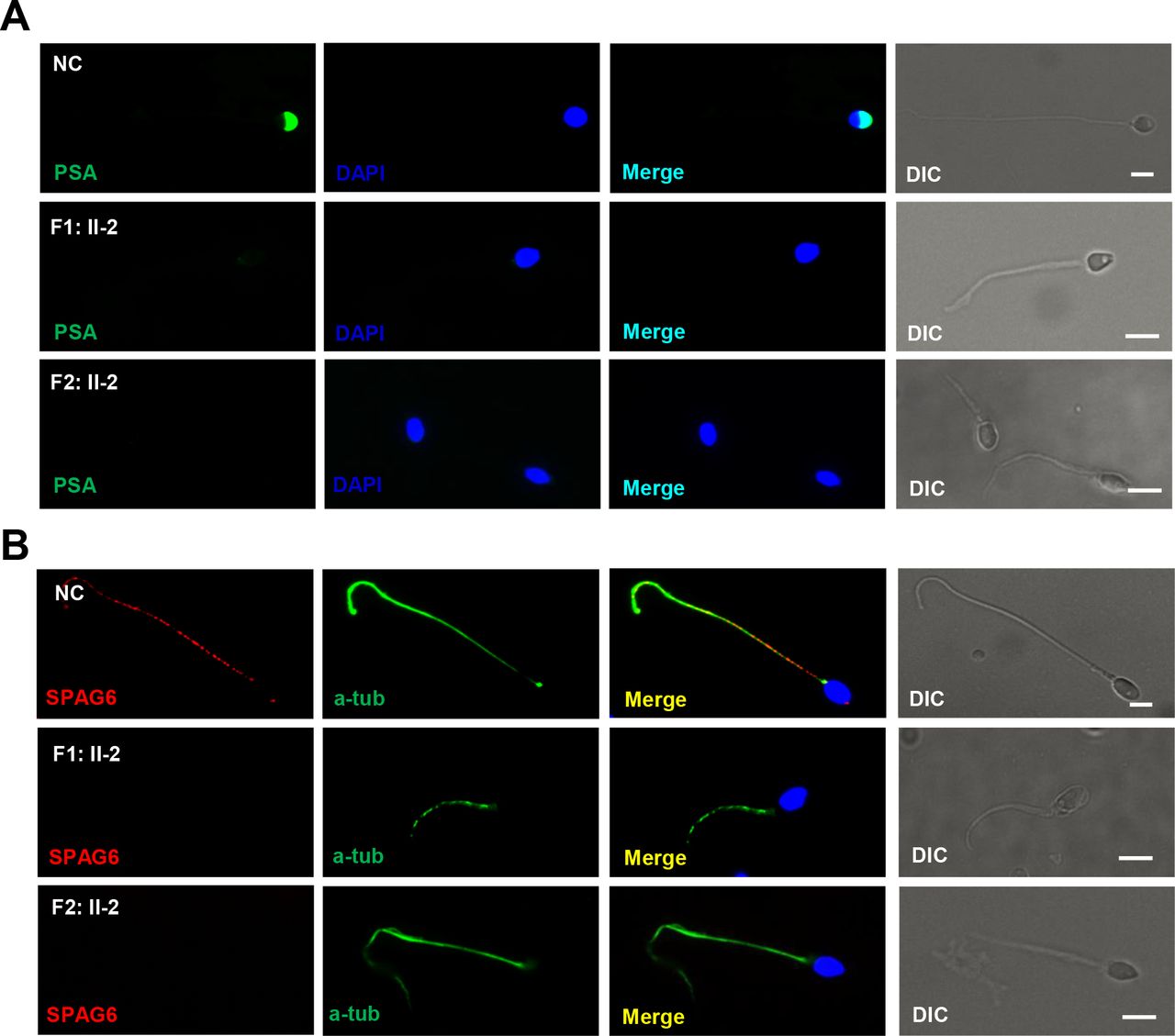
The location of CFAP65 in spermatozoa as well as ultrastructure analysis indicates that CFAP65 may play an essential role in acrosome formation and flagellum assembly. To investigate the effect of CFAP65 on acrosome formation and flagellum assembly, we used PSA staining conjugated to fluorescein isothiocyanate for evidencing the acrosome and performed immunostaining for SPAG6 (a CPC protein). PSA could bind glycoprotein in the acrosome, but its signal for acrosome was absent in 82.0% (41/50) and 77.6% (45/58) of the spermatozoa from F1: II-2 and F2: II-2. And abnormal shapes of the PSA signal were present in the rest of the spermatozoa from F1: II-2 and F2: II-2 in contrast to those of the normal controls (figure 5A and online supplementary figure S4). SPAG6 expression was absent in the axonemal components (figure 5B), which is consistent with the TEM results for CFAP65 mutant spermatozoa (figure 3). These results demonstrate that CFAP65 plays a key role in spermiogenesis by upholding the integrity of acrosomal and flagellar components.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The expression patterns of acrosome and CPC are changed in CFAP65 mutant spermatozoa. (A) PSA staining (green) conjugated to fluorescein isothiocyanate for evidencing the acrosome. Signals for acrosomes were absent in spermatozoa from F1: II-2 and F2: II-2 in contrast to those of NC. (B) Compared with NC, SPAG6 staining for CPC were absent in the sperm flagella of F1: II-2 and F2: II-2. Scale bars represent 5 µm. CPC, centralpair complex; DAPI, 2-(4-amidinophenyl)-1H-indole-6-carboxamidine; NC, normal control; PSA, Pisum sativum agglutinin; DIC, differential interference contrast.
ICSI outcomes for CFAP65 mutant individuals
All these three affected couples, including F1: II-2, F2: II-2 and F3: II-1, had accepted ICSI treatment in our hospital. However, as ICSIs had been conducted prior to the identification of CFAP65 mutations in males, mutation screening of CFAP65 was not performed for the female partners at that time. We then performed retrospective analyses for the ICSI outcome of these three couples (online supplementary table S5). The mean treatment age was 33.7, ranging from 26 to 39 years, for males, and 28.7 years, ranging from 24 to 37 years, for females. Each couple had undergone two ICSI cycles, but clinical pregnancy was not achieved. Fertilisation rates ranged from 22.2% (2/9) to 69.2% (9/13). In one treatment cycle for couple 1 (F1: II-2), two treatment cycles for couple 2 (F2: II-2) and two treatment cycles for couple 3 (F3: II-1), biochemical pregnancies were confirmed following the transfer of embryos, but no fetal heartbeat was detected via ultrasound and the pregnancies ended in abortion. Thus, findings related to the three patients with acrosomal hypoplasia and other patients with completely immotile spermatozoa who displayed a clinical pregnancy rate over 60%21 indicate that CFAP65 mutants may impede ICSI outcomes.
Discussion
To the best of our knowledge, the current study, for the first time, demonstrated that the presence of biallelic loss-of-function mutations of CFAP65 may induce severe defects in the tail and head of human spermatozoa, thus establishing CFAP65 as a candidate disease-causing gene for male infertility with completely immotile spermatozoa. CFAP65 could localise at the acrosome region and the flagellar midpiece of spermatozoa, possibly playing an important role in flagellum assembly and acrosome formation.
CFAP65 located on 2q35 was previously identified in human spermatozoa via mass spectrometry.22 The protein encoded by this gene is speculated to be a transmembrane protein containing two putative domains, ASH and MSP. The ASH domain of abnormal spindle-like microcephaly-associated protein has been found in proteins associated with cilia, flagella, centrosome and the Golgi complex. The MSP domain, which mainly functions as a protein–protein interaction module, performs other functions related to the formation of the dynamic MSP cytoskeleton and serves as a signalling molecule.23 The putative orthologue of CFAP65 in Chlamydomonas is strongly induced during the regeneration of the flagella.22 The effect of CFAP65 disruption on sperm motility was first discovered in Rosecomb chicken, where a 7.4 Mb inversion was present on chromosome 7. Two genes located at the inversion breakpoint, MNR2 and CFAP65, were suggested to cause the Rosecomb phenotype and defective sperm motility, respectively.24 Transcriptional analysis indicated that mitochondrial oxidation–reduction reactions were significantly downregulated in the testes of Rosecomb chicken,25 suggesting a potential role for CFAP65 in mitochondrial function.
In our study, although sperm acrosome may show possibly unspecific staining to PSA or CFAP65, both the ultrastructure analysis of spermatozoa and the immunostaining in testis sections could support the specific localisation of CFAP65 in the acrosome. The sperm phenotypes of two affected individuals with CFAP65 loss-of-function mutations, F1: II-2 and F2: II-2, were also systematically analysed. Prominent acrosome and MS defects, which resemble phenotypes observed in various murine models with acrosome biogenesis, such as those with deficiency in Hrb, Gopc, Spaca, Pick and so on, were revealed.7 ,26 However, the observed phenotypes were different from those in currently reported globozoospermia patients with PICK1, SPATA16 and DPY19L2 defects,10 ,12 such as in round-headed spermatozoa without acrosomes. As previously described, acrosomal defects observed in this study were most associated with acrosomal hypoplasia,8 ,9 suggesting a different role for CFAP65 in acrosome genesis.
CFAP65 belongs to the cilia- and flagella-associated protein (CFAP) family. Recently, mutations in four genes which belong to this family, including CFAP43 (MIM: 617558), CFAP44 (MIM: 617559),27 28 CFAP69 (MIM: 617949)29 and CFAP251 (MIM: 618146),30 have been identified as causing MMAF, a mosaic morphological abnormality in sperm flagella leading to absent, short, bent, coiled and irregular flagella. The flagellar phenotypes of spermatozoa in CFAP65 mutant individuals resemble those observed in patients with MMAF. However, CFAP65 mutant individuals also exhibited acrosomal abnormalities in the sperm head, which have not been extensively studied in patients with MMAF. Therefore, phenotypes caused by CFAP65 mutations may be a phenotypic variation of classical MMAF. Moreover, two of the affected individuals, F1: II-2 and F2: II-2, in our study exhibited typical PCD symptoms although a definitive diagnosis of PCD was not confirmed. Considering CFAP65 expression in some ciliated tissues, the possibility of PCD with weaker symptoms resulting from a CFAP65 defect cannot be ruled out. Further studies may be needed to reveal CFAP65 involvement in ciliary function.
The current study indicated that CFAP65 was expressed in the flagellar midpiece and the acrosome of mature spermatozoa. Mitochondria and acrosome are organelles containing various enzymes and are the main Ca2+ storage sites.31 A previous study demonstrated that CFAP65 was involved in calcium-mediated retrograde signalling affecting cell differentiation and proliferation, initiated by the depletion of mitochondrial DNA.32 Based on current evidence, we speculate that CFAP65 may, on one hand, participate in calcium-mediated activities, and on the other hand mediate the anchoring of mitochondria/acrosomes to the outer dense fibres/nuclear membrane forming the cytoskeleton.33 34 As there is excess calmodulin in the CPC35 36 and Ca2+-binding regulated protein in the FS,37 the absence of CPC and disorganisation of FS may be a secondary defect caused by the dysregulation of CFAP65-mediated activities. Thus, further investigations may be needed to reveal functional network of CFAP65 as well as mechanisms underlying CFAP65 function in spermiogenesis.
ICSI is a solution suggested for treating individuals with severe asthenoteratospermia, but the fertility prognosis in acrosome-defect cases was comparatively lower than that of other asthenozoospermia cases, such as PCD or MMAF.7 Additionally, the poor ICSI outcomes seen in our study suggest that CFAP65 deficiency may affect the sperm head, chromatin or the integrity of the sperm centrosome, which are essential for regulating syngamy and first mitosis following fertilisation, and thus, the developmental potential of the embryo.38 Recent studies associated with spermatozoa carrying acrosomal anomalies have attempted to activate oocytes using Ca2+ ionophore, strontium or electrical activation, resulting in pregnancies and live births.39–41 Considered together, mutation screening of CFAP65 as well as oocyte activation in female partners may be useful to improve ICSI success for males with biallelic CFAP65 mutations and reduce the potential risk to offspring. Moreover, due to the small size of the samples used, further investigation may be required to determine whether successful ICSI outcomes can be achieved in patients carrying CFAP65 mutations.
In conclusion, these findings which identified three biallelic mutations in CFAP65 may be expected to further knowledge on genetic pathogeny associated with male infertility due to severe asthenoteratospermia. This finding may also have important clinical implications for genetic and reproductive counselling of affected families. Deletion of CFAP65 would seriously impact acrosome formation and flagellum assembly. However, there were certain limitations in the present study. Further investigations are required to identify the functional role of CFAP65 in acrosome formation and flagellum assembly, and such investigations may involve the screening of a large number of patients with severe asthenoteratospermia and the generation of mouse knockout models using knock-in alleles that mimic mutations found in patients with severe asthenoteratospermia.
Acknowledgments
The authors thank all the families of the patients and the individuals who participated in this study. We acknowledge the support from the clinical and nursing staff at the Reproductive and Genetic Hospital of CITIC-Xiangya. We also thank Dr Xiaoyin Wu at the Xiangya Hospital for technical assistance with SEM.
References
Footnotes
Contributors Y-QT designed the research; WW, CT performed the research; HN and YL collected the samples of patients; JW performed the TEM analysis; LM, SY, QZ and JD performed the bioinformatics analysis; FG, G-XL, LF and GL analysed the data; Y-QT, WW and CT analysed the data and wrote the paper.
Funding This study was supported by grants from the National Key Science Program S&T Program (2018YFC1004900 to Y-QT), the National Natural Science Foundation of China (81771645 to Y-QT), the National Key Research and Development Program of China (2016YFC1000206 to GL), the science and technology major project of the Ministry of Science and Technology of Hunan Province (2017SK1030 to Y-QT), the Graduate Research and Innovation Projects of Central South University (grant 2019zzts322 to WW).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The institutional ethics committees of the Central South University and Reproductive Genetic Hospital of CITIC Xiangya.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.