Article Text

Abstract
Background Mutations in the PCDH19 gene have mainly been reported in female patients with epilepsy. To date, PCDH19 mutations have been reported in hundreds of females and only in 10 mosaic male epileptic patients with mosaicism.
Objective We aimed to investigate the occurrence of mosaic PCDH19 mutations in 42 families comprising at least one patient with PCDH19-related epilepsy.
Methods Two male patients with mosaic PCDH19 variants were identified using targeted next-generation sequencing. Forty female patients with PCDH19 variants were identified by Sanger sequencing and Multiple Ligation Probe Amplification (MLPA). Microdroplet digital PCR was used to quantify the mutant allelic fractions (MAFs) in 20 families with PCDH19 variants.
Results Five mosaic individuals, four males and one female, were identified in total. Mosaic variant was confirmed in multiple somatic tissues from one male patient and in blood from the other male patient. Among 22 female patients harbouring a newly occurred PCDH19 variant identified by Sanger sequencing and MLPA, Sanger sequencing revealed two mosaic fathers (9%, 2/22), one with two affected daughters and the other with an affected child. Two asymptomatic mosaic fathers were confirmed as gonosomal mosaicism, with MAFs ranging from 4.16% to 37.38% and from 1.27% to 19.13%, respectively. In 11 families with apparent de novo variants, 1 female patient was identified as a mosaic with a blood MAF of 26.72%.
Conclusion Our study provides new insights into phenotype-genotype correlations in PCDH19 related epilepsy and the finding of high-frequency mosaicism has important implications for genetic counselling.
- epilepsy
- mddpcr
- mosaicism
- pcdh19
- sequencing
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Point mutations or rearrangements involving the PCDH19 gene (MIM# 300460) located on Xq22.1 cause epilepsy and mental retardation mainly in females.1–3 The main clinical manifestations of PCDH19-related epilepsy include early seizure onset, generalised or focal seizures highly sensitive to fever and brief seizures occurring in clusters.4 The disorder shows an unusual X-linked inheritance affecting heterozygous females and sparing hemizygous males.1 However, males with mosaic mutations can also be affected.5 This mode of inheritance might be explained by ‘cellular interference’.5 6 According to this hypothesis, the coexistence of wild-type and mutant cell populations may scramble cell-cell communication.
Recently, next-generation sequencing (NGS) technologies have enabled the identification of mosaicism for PCDH19 point mutations in male patients with epilepsy.7 8 To date, PCDH19 mutations have been reported in hundreds of females and only in 10 mosiac male patients with epilepsy.5–9 In addition, mosaic PCDH19 mutations have been reported in mildly affected or even unaffected mothers.10 11 Interestingly, we identified an asymptomatic mosaic father with two affected daughters.12 Our previous study revealed parental mosaicism for approximately 10% of apparent de novo SCN1A mutations in children with Dravet syndrome.13 Since de novo PCDH19 mutations occurred in more than half of the sporadic cases,14 we suspected that parental mosaic mutations of PCDH19 might be underestimated.
In this study, we investigated 42 Chinese families comprising at least one patient with PCDH19-related epilepsy (online supplementary figure 1). Our objectives were to estimate the frequency of mosaicism in a Chinese cohort of PCDH19-related epileptic families and to determine the role of mosaicism in phenotypic variations, thereby providing better informed genetic counselling.
Supplementary file 1
Methods
Subjects
We recruited 42 PCDH19-related epileptic families from the Pediatric Clinic of Peking University First Hospital and Wuhan Children’s Hospital between October 2007 and July 2017 (online supplementary figure 2). Written informed consent was provided by participants or their statutory guardians before enrolment. In the 42 families, 21 families were previously reported12 and 21 were families newly recruited. The clinical features and the results of the mutation analysis in these new families are summarised in table 1.
The clinical manifestations of 21 newly assessed patients with epilepsy with PCDH19 pathogenic variants
Genetic analysis
Peripheral blood leucocytes obtained from the 42 families, as well as samples obtained from other available tissues, were used for this study. Genomic DNA from peripheral blood was isolated using a simple salting-out procedure.15 Genomic DNA from saliva, hair follicles, buccal swab, urine and finger nails of four individuals with suspected mosaicism was collected using a QIAamp DNA micro kit (Qiagen) or a TIANamp micro DNA kit (Tiangen).13 16 A PureSperm 40/80 assay (Nidacon) was used for the purification of vital sperm from paternal semen sample, and DNA was extracted from purified sperm using a phenol-chloroform extraction method.
Two male patients with mosiac PCDH19 variants were ascertained by clinical testing, both with epilepsy and intellectual/development disabilities. For male patient 25, a custom-designed panel for capturing the coding exons of 470 genes associated with epilepsy,17 including SCN1A and PCDH19 (NM_0011848801.1), based on the Agilent SureSelect Target Enrichment technique (Zhongguancun Huakang Gene Institute, China) was used. For male patient 26, another custom-designed panel capturing the coding exons of 153 genes associated with epilepsy (online supplementary table S1), including SCN1A and PCDH19, were synthesised by Agilent Technologies on a chip (MyGenostics, Baltimore, Maryland, USA). Targeted gene capture, massively parallel sequencing and sequence alignment were performed for the two epileptic and intellectual disability patients as described previously.17 18 All sequencing data were based on the reference human genome build hg19.
PCDH19 point variants were detected by PCR and Sanger sequencing as previously described.12 SALSA Multiple Ligation Probe Amplification (MLPA) probe mix P330-A2 was applied to confirm PCDH19 rearrangements. Segregation analysis was performed for all available members of a pedigree. If neither parent had the variant, then parent-offspring trios were tested by microsatellite markers analysis at the X chromosome to ensure that the variant occurred de novo.
Quantification of mutant versus wild-type allele
Customised TaqMan SNP Genotyping Assays (Life Technologies, Grand Island, New York, USA), consisting of mutation-specific primers and fluorescent-labelled allele discrimination probes, were used to quantify the mutant allelic fraction (MAF) of each site measured using the RainDance Raindrop microdroplet digital PCR platform (mDDPCR, RainDance, Billerica, Massachusetts, USA). mDDPCR was performed in 20 families with PCDH19 variants (including four families with a male mosaicism, 11 parent-offspring trios with apparent de novo heterozygous mutations in probands by Sanger sequencing, 3 families with mild affected or asymptomatic mothers and 2 families with hemizygous fathers), as previously described.13
Mosaic individuals were normalised to the normal control identified in the same family. In families with a mosaic or hemizygous father, we also controlled for PCR efficiency by assuming that 100% of the affected offspring’s cells harboured a mutation.
Results
PCDH19 mutations in 42 families with epilepsy
By the end of July 2017, a total of 42 probands with epilepsy were identified to carry PCDH19 pathogenic variants, including 40 females and 2 mosaic males. Forty of the probands exhibited point variants (21 previously reported) and two (2/42, 5%) probands demonstrated whole-gene deletion of PCDH19. We found 37 different pathogenic variants, including 15 missense variants, 12 small frameshift insertions and deletions (indels), 7 nonsense variants, 1 small in-frame insertion, 1 splicing variant and 1 whole gene deletion. In 21 newly described probands, we identified 12 novel and 9 reported variants (table 1). All four novel missense variants were on exon 1 and were predicted as pathogenic using the in silico prediction tools Mutation Taster Server, Polyphen-2 and SIFT. None of the novel mutations was detected in 100 Chinese healthy controls, the dbSNP or ExAC databases.
Among the 40 female patients, 19 patients were identified with inherited variants and 21 patients demonstrated apparent de novo variants by Sanger sequencing and MLPA. In our 40 female proband families, 60 females were confirmed to carry PCDH19 variants and 54 of had an epileptic seizure. Thus, the penetrance of females with the PCDH19 variants was estimated as 90% (54/60) in our study. Twelve hemizygous fathers and the previously reported mosaic father (patient 13’s father) were asymptomatic.12
mDDPCR analysis was performed in 20 families with PCDH19 pathogenic variants, involving 65 participants, of which 23 were males and 42 were females (including 4 families with a male mosaicism, 11 parent-offspring trios of which the probands were detected with seemingly de novo variants, 3 families with mild or asymptomatic mothers and 2 families with hemizygous fathers). The data are shown in online supplementary table S2. To evaluate the accuracy of the MAFs quantified by mDDPCR, we screened two fathers with hemizygous PCDH19 variants as controls. The MAF was measured as 99.69% (95% binomial CIs 99.58% to 99.77%) and 99.95% (95% binomial CIs 99.88% to 99.99%). In addition, the 95% CI of MAFs measured in eight females with an inherited variant ranged from 44.53% to 55.17%.
Mosaicism in male patients
Two male mosaic patients (Probands 25 and 26) with epilepsy demonstrated intellectual disability. Proband 26 was also diagnosed with autism spectrum disorder. They were identified by targeted NGS. A total of 189 males with epilepsy have been screened using gene-panel testing from 2014 to 2017, only two (2/189, 1%) were identified with PCDH19 variants. In patient 25, the c.317T>A (p.Met106Arg) variant in PCDH19 was preliminary detected as a hemizygous mutation. Manual inspection revealed 85% of the mutant allele in an 86X coverage region (MAF 85.0%). Sanger sequencing confirmed the existence of PCDH19 mosaic variants in his blood, buccal epithelium samples. It also suggested the existence of PCDH19 mosaic variant in his saliva and urine samples. However, the variant was present in almost 100% of the hair follicles because it appeared in a hemizygous status (figure 1A). In patient 26, the c.158dupT (p.Asp54GlyfsX35) variant was detected as a mosaic mutation by NGS. It revealed 33% of the mutant allele in a 124X coverage region (MAF 33.1%). We sequenced every mosaic variant bidirectionally, for at least twice, in technical replicates starting from independent PCR reactions. Segregation analysis demonstrated that these two variants occurred de novo. MLPA analysis did not detect any PCDH19 deletion/duplication in the two male patients.
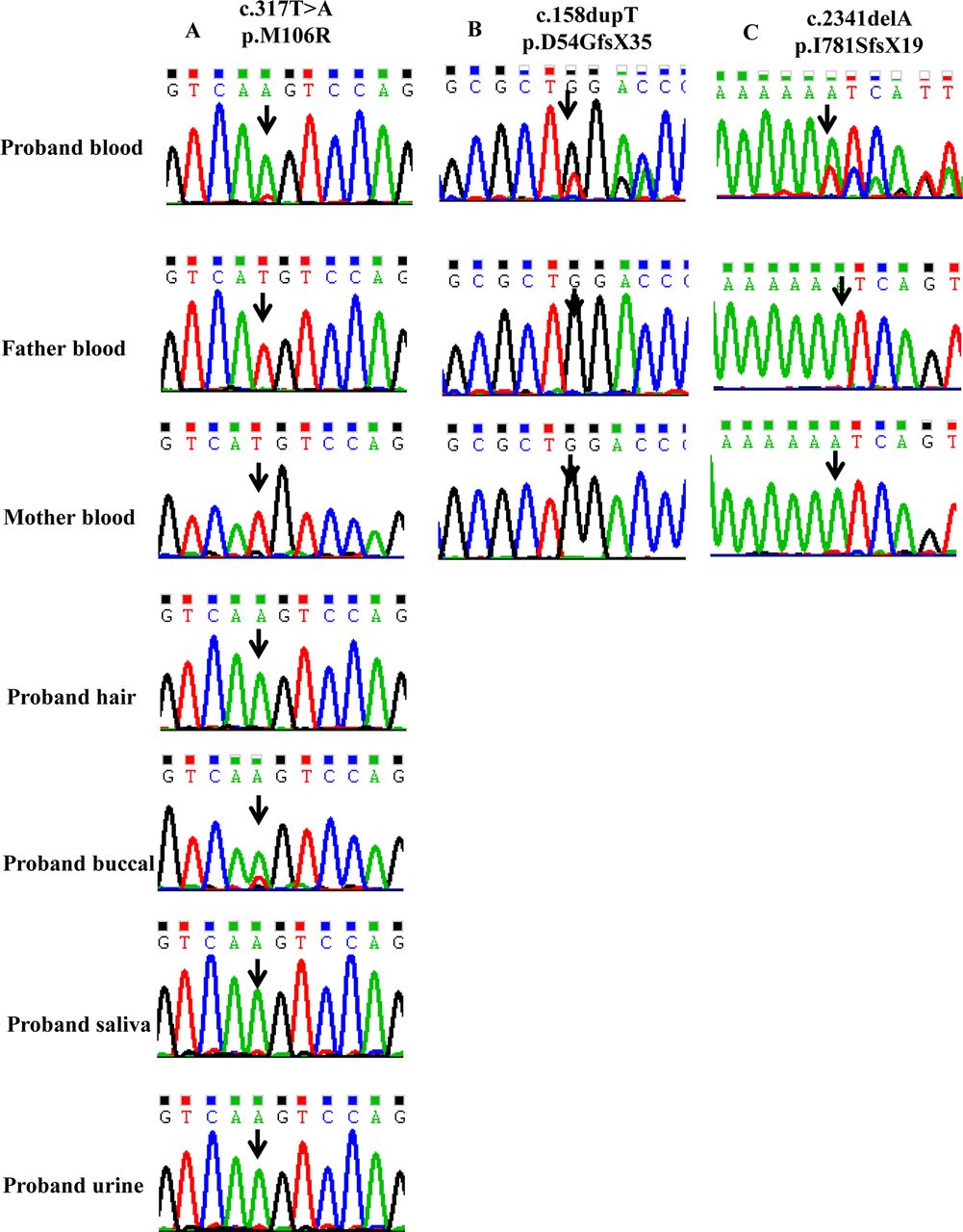
Sanger sequencing chromatograms of three mosaic probands families. (A) Sanger sequencing shows that the proband is mosaic for the PCDH19 variant (c.317T>A) in blood, buccal epithelium, saliva and urine. The variant appears to be hemizygous in his hair follicles. The variant is absent in his parents’ blood. (B) Sanger sequencing shows that the proband is mosaic for the PCDH19 variant (c.158dupT) in blood. The variant is absent in his parents’ blood. (C) Sanger sequencing shows that the proband is mosaic for the PCDH19 variant (c.2341delA) in blood. The variant is absent in her parents’ blood.
We confirmed and quantified the MAFs in the two male mosaic cases by mDDPCR. The MAFs of male patient 25’s multiple somatic tissues (blood, hair follicles, buccal swab, finger nails, saliva and urine) were 67.43%–98.46% (figure 2). The MAF of male patient 26’s blood DNA was 37.08%.

The mDDPCR results of Family 25. Detection of PCDH19 c.317T>A mutation in Proband 25’s multiple tissues (blood, buccal epithelium, saliva and urine). The wild-type and mutant population are circled in the top panels and bottom panels, respectively, with the % MUT indicated in the top right corner (MUT drops/total of WT+MUT droplets). MAF, mutant allelic fraction; mDDPCR, microdroplet digital PCR; WT, wild type.
Patient 25 was 3 years old at last follow-up. He was born full term to a G2P2 34-year-old mother. His birth weight was 3.25 kg. Both the pregnancy and neonatal course were unremarkable. Familial and personal neurological antecedents were negative. However, his mother was diagnosed with a teratoma on a physical examination at the age of 29 years old between her two pregnancies. The patient developed seizures at 5 months of age lasting 20–50 s. A cluster of attacks occurred every 1–2 months, with each cluster lasting for 1–15 days. He has predominantly afebrile focal tonic seizures. Antiepileptic drugs were arbitrarily stopped after he had been treated with oxcarbazepine, levetiracetam and valproate without effect. He tended to display excessive motor activity and was described as restless. His cranial MRI was normal. He has no other significant medical concerns. His psychomotor development, prior to the onset of epilepsy, was reported as normal. He manifested severe cognitive impairment after seizure onset. An interictal EEG (while medicated with oxcarbazepine and levetiracetam) at 9 months was normal. An EEG (2 days after seizures cluster) at 1 year was abnormal, demonstrating epileptic discharges in the bilateral occipital and temporal posterior regions. An EEG (during the time of the seizure cluster) at 13 months recorded dozens of focal seizures from the anterior region.
Patient 26 was 9 years old at last follow-up. He was born 37+4 to a G1P2 mother. His birth weight was 2.3 kg. Both the pregnancy and neonatal course were unremarkable. Familial and personal neurological antecedents were negative. At the age of 9 months, he presented the first cluster of febrile seizures. Multiple seizure types occurred including febrile and afebrile generalised tonic-clonic seizures, myoclonic seizures and absence seizures. Seizures were highly sensitive to fever. He had been treated with phenobarbital and subsequently with valproate and topiramate. His seizures were controlled by valproate and topiramate since he was 4 years old. He had autistic traits, excessive motor activity and was described as restless. His cranial MRI was normal. He had no other significant medical concerns. An EEG (while medicated with valproate and topiramate) at 2 years and 10 months was abnormal, showing diffuse slowing of background activity, epileptic discharges in the bilateral, frontal and central regions, evident during drowsiness.
Mosaicism in the female patient
In 11 families in which the probands with apparent de novo variants by Sanger sequencing, 1 female patient (female patient 29) was identified as a mosaic with a blood MAF of 26.72% (figure 1C). No parental mosaicism was identified in these 11 families.
Patient 29 was 2 years at the last follow-up. She was born full term. Both the pregnancy and neonatal course were unremarkable. She had a history of febrile seizure at the age of 12 months. At the age of 15 months, she presented the first cluster of febrile seizures. She has predominantly generalised tonic-clonic seizures and focal seizures. Her seizures were highly sensitive to fever. Her cranial MRI was normal. She had no other significant medical concerns. Her EEG at 15 months captured six seizures, which manifestated staring blankly, head deviation to the back and tonic in four limbs. The EEG was abnormal, showing diffuse slowing of background activity and epileptic discharges in the bilateral regions, predominantly in the left hemisphere. She had been treated with levetiracetam since 15 months. She had two more clusters of seizures during the next year. She had normal development.
Parental mosaicism
Since technical limitations and manual limitations are the frequent reasons for missed SCN1A mutations (including heterozygous and mosaic mutations),19 we carefully reconfirmed the Sanger sequencing results in the 19 families, which were initially assumed as de novo PCDH19 point variants. Consequently, another asymptomatic mosaic father (patient 1’s father) was identified, as shown in figure 3A and B. Sanger sequencing confirmed the PCDH19 mosaic variants in Father 1’s blood, hair follicles, buccal swab, finger nails, saliva, urine and purified sperm. Sanger sequencing confirmed PCDH19 mosaic variants in Father 13’s blood, buccal swab, finger nails, saliva and sperm. However, the variant was absent in his urine. Thus, the two fathers were confirmed as gonosomal mosaicism. In addition, the two mosaic fathers identified by Sanger sequencing were confirmed as a somatic (blood, hair follicles, buccal swab, finger nails, saliva and urine) and gonadal mosaicism by mDDPCR, with MAFs of 4.16%–37.38% and 1.27%–19.13%, respectively, (figure 4 and figure 5).
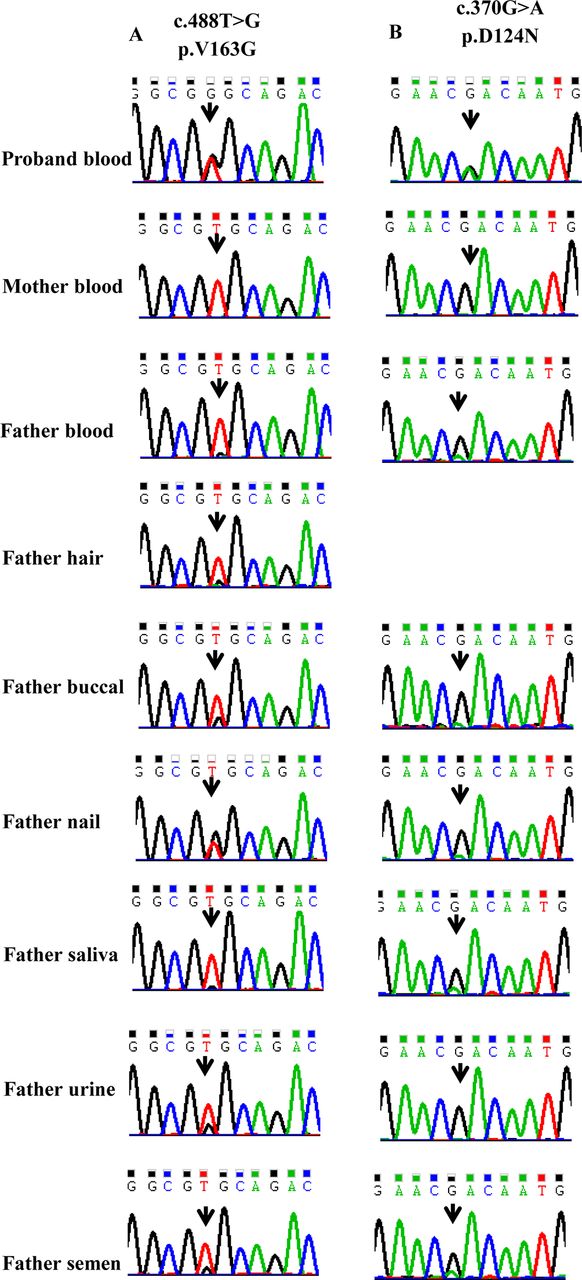
Sanger sequencing chromatograms of two parental mosaic families. (A) Sanger sequencing shows that the proband is heterozygous for the PCDH19 variant (c.488T>G) and her father is mosaic for the same variant in blood, hair, buccal epithelium, finger nails, saliva, urine and purified sperm. The variant is absent in her mother’s blood. (B) Sanger sequencing shows that the proband is heterozygous for the PCDH19 variant (c.370G>A) and her father is mosaic for the same variant in blood, buccal epithelium, finger nails, saliva, urine and purified sperms. The variant is absent in her mother’s blood.

The mDDPCR results of Family 1. Detection of PCDH19 c.488T>G mutation in Proband 1’s blood and her father’s multiple tissues (blood, hair follicles, buccal epithelium, finger nails, saliva, urine and purified sperms). The wild-type (‘WT’) and mutant (‘MUT’ or ‘MU’) population are circled in the top panels and bottom panels, respectively, with the % MUT indicated in the top right corner (MUT drops/total of WT+MUT droplets). MAF, mutant allelic fraction; mDDPCR, microdroplet digital PCR; WT, wild type.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The mDDPCR results of Family 13. Detection of PCDH19 c.370G>A mutation in Proband 13’s blood, her sister’s blood and their father’s multiple tissues (blood, buccal epithelium, finger nails, saliva, urine and purified sperms). The wild-type and mutant population are circled in the top panels and bottom panels, respectively, with the % MUT indicated in the top right corner (MUT drops/total of WT+MUT droplets). MAF, mutant allelic fraction; mDDPCR, microdroplet digital PCR; WT, wild type.
The clinical details of Family 1 and Family 13 have been reported. Proband 1 is a sporadic case (patient 1 in Liu et al 2017).12 Proband 13 has an affected sibling (patient 13 in Liu et al 2017).12
Incomplete penetrance in heterozygous females is unrelated to mosaicism
We further investigated whether mosaicism plays a role in females with a mild phenotype or those who are asymptomatic. The mother of patient 4 had febrile and afebrile seizures before 13 years of age and exhibited normal intelligence. The mother of patient 6 had only febrile seizures in her childhood. The MAFs from multiple somatic tissues (blood, hair follicles, buccal swab, saliva and urine) in patient 14’s mother ranged from 47.20%–49.64%. On the basis of these data, these patients were determined not to exhibit somatic mosaicism.
Discussion
Mosaicism is a common biological phenomenon. It describes an individual who has developed from a single zygote and has two or more populations of cells with distinct genotypes.20 Mosaicism can occur at any time during development after the formation of the fertilised egg.21 It is known that somatic mutations have important roles in cancer22 23 and monogenic diseases.24–30 In monogenic diseases, if mutations occur in the early embryo stage, the individual may be composed of mixed gonadal and somatic mosaicism.31 Somatic and gonadal mosaicism in the parents can cause recurrent transmission of human genetic disease.31 32 This phenomenon has been reported in more than 100 genes, such as DMD, SCN1A, ACTA1 and so on.24 26 33 Somatic mosaicism can also contribute to variable phenotypic expressivity.33 34 For the purpose of this study, we focused on the somatic mosaicism in PCDH19.
Until recently, 10 male mosaic epilepsy patients with PCDH19 mutations have been reported.5–9 In this study, we identified two new male mosaic epilepsy patients with PCDH19 mutations by NGS. Previously, male epilepsy patients with mosaic PCDH19 mutations were explained by the ‘cellular interference’ mechanism because the coexistence of wild-type and mutant cell populations may scramble cell-cell communication.5 However, we identified an asymptomatic mosaic father who had two affected daughters.12 Apparent de novo mutations may actually be inherited from mosaic parents.13 However, mutations in mosaic parents could be undetectable due to technical limitations. We reanalysed the Sanger sequencing results of 19 families in which the probands were initially considered as de novo PCDH19 point mutations. The second asymptomatic mosaic father, who was initially overlooked, was found in families which had one child with apparent sporadic PCDH19-related epilepsy. Our identification of the two asymptomatic mosaic fathers implies that the frequency of paternal mosaicism might be underestimated. The identification of asymptomatic mosaic fathers is inconsistent with previous reports, in which mosaic males have clinical manifestations.5–9 We hypothesised that the ratio of mutant versus wild-type protocadherin 19 protein in the brain determines the phenotypes in mosaic males. To test this hypothesis, we quantified the mosaic ratio at the PCDH19 locus using mDDPCR. The MAFs in the two mosaic male patients were 37.08%–98.46%. The mutant allele fractions in the two asymptomatic fathers were 4.16%–37.38% and 1.27%–19.13% in different tissues. In our cohort, the affected males have a higher quantity of mutations than the unaffected fathers, although the percentage of mutation varies from one tissue to the other. The blood MAFs in the two patients was closer to 50% compared with the two asymptomatic fathers. So we suspect that a male with a mutant percentage near to 50% in the brain seems more likely to be affected, while males with too high or too low percentage of mosaicism may not be affected. Although we assumed the clinical outcome of mosaic males was related to the mosaic MAFs, we could not identify a definite cut-off value. In Terracciano’s two male patients,7 the mosaic ratio was 10% and 90%, but in our cohort, the two asymptomatic fathers with 13% and 19% mosaicism in blood. However, the percentage of mosaicism in the brain cannot be extrapolated from the blood.5
Maternal mosaicism of PCDH19 mutations has also been previously reported.10 The mosaic mothers were affected or asymptomatic due to random X inactivation or the ratio of mutant versus wild-type protocadherin 19 protein. Thus, parental mosaicism should be considered when providing genetic counselling for couples who have one affected offspring. We successfully found two asymptomatic mosaic fathers by Sanger sequencing; therefore, the parents’ Sanger sequencing results of other cohorts should be reanalysed to avoid negligence of mosaic situations.19 However, Sanger sequencing could not detect mosaicism with a MAF <5% and could hardly distinguish heterozygous mutations from mosaic mutations in females. Mosaicism is now more readily detectable and quantified by mDDPCR or other deep sequencing techniques in families with assumed de novo mutations by Sanger sequencing.13 NGS is a powerful tool for mosaicism detection. Among the 14 males (10 males previously reported and 4 males reported in this study, online supplementary table S3) with somatic mosaic mutations in PCDH19, 9 (64%) were detected by targeted NGS. To quantify the mosaic ratio, we applied the more sensitive mDDPCR method which could detect MAF as low as 0.01% in our study.13
Somatic gene conversion from wild-type to mutant is known as ‘forward mosaicism’.35 On the other hand, spontaneous gene correction can happen in the opposite direction during mitosis, known as ‘revertant mosaicism’.35 Back mutation now becomes an important genetic mechanism to consider when explaining examples of a reversion of somatic cells to ‘normal’ in persons with a genetically determined abnormal phenotype.36 In our study, male patient 25 had a mutant allele of more than 50%. As one male’s PCDH19 allele was inherited from his mother, the mutation was assumed to already be present in the oocyte prior to fertilisation. Back mutation was suspected to occur during the early development in this male patient, leading to postzygotic PCDH19 mosaicisms. This suspected reversion mechanism may be a reason for male patients with mosiac PCDH19 mutations. By contrast, spontaneous genetic reversion has been described as ‘natural gene therapy’ in some disease, such as Fanconi anaemia and dyskeratosis congenita.37 38 In our study, the mother of patient 25 with PCDH19 c.317T>A gonadal mosaicism had developed a teratoma. In a previous study, the PCDH19 c.918C>T mutation found in a male mosaic patient had been previously reported in somatic tumour tissue.7 Based on these findings, tissue limited mosaicism for PCDH19 mutations may have a relationship with tumours and thus demand special attention.
Moreover, we identified one mosaic female proband in 11 families with apparent de novo PCDH19 mutations using mDDPCR. Mosaicism can also be an important cause of phenotypic variation in some females.11 We quantified three mildly affected or asymptomatic mothers in our cohort using mDDPCR. The mutant allele ratio was all nearly 50% and was thus ruled out for mosaicism. This finding suggests other genetic or non-genetic modifiers, rather than mosaicism, which may also be involved in the phenotype of females, such as X inactivation.39
Overall five mosaics, including two male patients and one female patient and two asymptomatic mosaic fathers were identified in our PCDH19 gene study. This finding further demonstrated that PCDH19 mutations could occur in males and females at any development stage. The observation of asymptomatic mosaic fathers suggested that the frequency of parental mosaicism is underestimated. Taken together, our data implied that parental mosaicism should be considered during genetic counselling. We hypothesised that the mosaic MAFs in males may be related to phenotype, although other genetic or non-genetic modifiers may also be involved.
Acknowledgments
The authors thank the patients and their family members for participating in this study. The authors would like to thank Beijing Key Laboratory of Molecular Diagnosis and Study on Pediatric Genetic Diseases for its help.
References
Footnotes
AL, XxY and XIY contributed equally.
Contributors AL and XxY prepared the manuscript. AL, XlY and JZ performed the Sanger sequencing and MLPA. XxY and QW performed microdroplet digital PCR. DS, ZY, YJ, XW and YZ conducted sample acquisition and collected clinical information. LW and YZ designed the study and edited the manuscript.
Funding This study was supported by grants from the National Key Research and Development Program of China (No. 2016YFC0904401 and 2016YFC0904400); National Natural Science Foundation for Young Scientists of China (No. 81701274), National Natural Science Foundation of China (No. 31530092) and Peking University Clinical Cooperation ‘985 Project’ (No. PKU-2014-1-1).
Competing interests None declared.
Patient consent Parental/guardian consent obtained.
Ethics approval Institutional Review Board at Peking University (IRBPU), the Ethics Committee of Peking University First Hospital and the Ethics Committee of Wuhan Children’s Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.