Article Text

Statistics from Altmetric.com
Partial trisomy or tetrasomy of the short arm of chromosome 9 are among the most common autosomal structural chromosomal anomalies in humans, so the phenotype-genotype correlation of these aneusomies has been well described. Characteristic clinical features of partial trisomy 9p are mental retardation of various degree, short stature, craniofacial abnormalities, short fingers, simian crease, and single crease of the fifth finger. Additional symptoms like microcephaly, cleft lip and palate, malformed ears, and skeletal, nail, cardiac, and genital anomalies have also been observed.1,2 In 1970, the first case of trisomy 9p was reported by Réthoré et al.3 Since then, more than 150 patients with partial or complete trisomy 9p have been reported. In most patients, the trisomic segment was transmitted from a parent carrying a reciprocal balanced translocation and only a small number arose from de novo duplications.
Here we report on a three generation family with an interchromosomal insertion of chromosome 9p12-p21 material into the short arm of chromosome 5. One member of the family carried a deletion in the inserted region resulting in cri du chat syndrome, whereas her father is trisomic for the inserted segment owing to an unbalanced segregation of the insertion chromosome. Surprisingly, the unbalanced insertion carrier does not show any morphological or mental abnormalities. The normal phenotype suggests that not all partial trisomies 9p are associated with clinical abnormalities. In particular, the proximal part of the short arm of chromosome 9 seems to be less important for the trisomy 9p phenotype.
CASE REPORTS
The female patient, III.1 (fig 1), is the third child of a 27 year old mother and the first child of a 24 year old father. The pregnancy and delivery were uneventful, the birth weight was 2830 g (3rd-10th centile), and length 48 cm (∼3th centile). The infant had feeding problems and chromosome analysis was performed because of clinical features of cri du chat syndrome, such as microcephaly, a round face, downward slanting palpebral fissures, deep set ears, microgenia, and atypical crying. Cytogenetic analysis showed the karyotype 46,XX,del(5)(p?).ish del(5)(p?)(D5S23−).

Truncated pedigree of the family, including the important data.
Because about 15% of cri du chat syndrome cases arise from parental chromosomal aberrations including reciprocal balanced translocations and inversions, chromosome analyses were also performed in the mother (II.1) and the father (II.2) of the affected child. The mother had a normal karyotype (46,XX), whereas a derivative chromosome 5 with an enlarged short arm was detected in the father (46,XY,der(5p)) (fig 2). Because the origin of the additional material could not be determined by GTG banding, molecular cytogenetic analyses (CGH) were initiated. Further chromosome analyses of the grandmother (I.2) and the uncle (II.3) of the index patient (III.1) were also performed.

GTG banded chromosomes 9 and 5 from a metaphase of II.2. Both chromosomes 9 and one chromosome 5 are normal. The der(5) harbours the unbalanced insertion of chromosomal bands (9)(p12p21.3).
II.2 is a healthy man without any morphological or mental abnormalities. He had normal stature, craniofacial habitus, and dermal ridges of the hands. There were no internal malformations. He had attended a secondary school and learned a technical profession.
METHODS
Metaphase chromosomes from PHA stimulated peripheral blood lymphocytes of the proband, his daughter, his mother, and his brother were analysed by standard GTG banding procedures and by FISH techniques. Appropriate informed consent was obtained from the family members. FISH studies were performed using the probe D5S23 (ONCOR), which is localised in the cri du chat syndrome critical region in 5p15.2, and the chromosome painting probes Coatasome 5 dig (ONCOR) and chromosome paint 9 bio (AGS) according to the supplier's protocol. CGH analysis was performed as previously described.4 Seven YAC clones from the short arm of chromosome 9p were selected from the website of the Molecular Cytogenetic and Positional Cloning Centre of the Max Planck Institute for Molecular Genetics (Berlin, Germany). YAC-FISH was performed as described by Stumm et al.5 Multicolour banding (mBand, Metasystems, Germany) analysis was carried out using seven region specific partial chromosome paints (RPCP) of chromosome 5, which were generated as described by Chudoba et al.6
RESULTS
Because the composition of the derivative chromosome 5 of II.2 could not be determined by GTG banding (fig 2), a whole chromosome painting (WCP) with a chromosome 5 specific probe was performed on metaphases of II.2. In the short arm of the derivative chromosome 5, a gap in the painting pattern was detected, indicating an insertion of chromosomal material from a non-homologous chromosome.
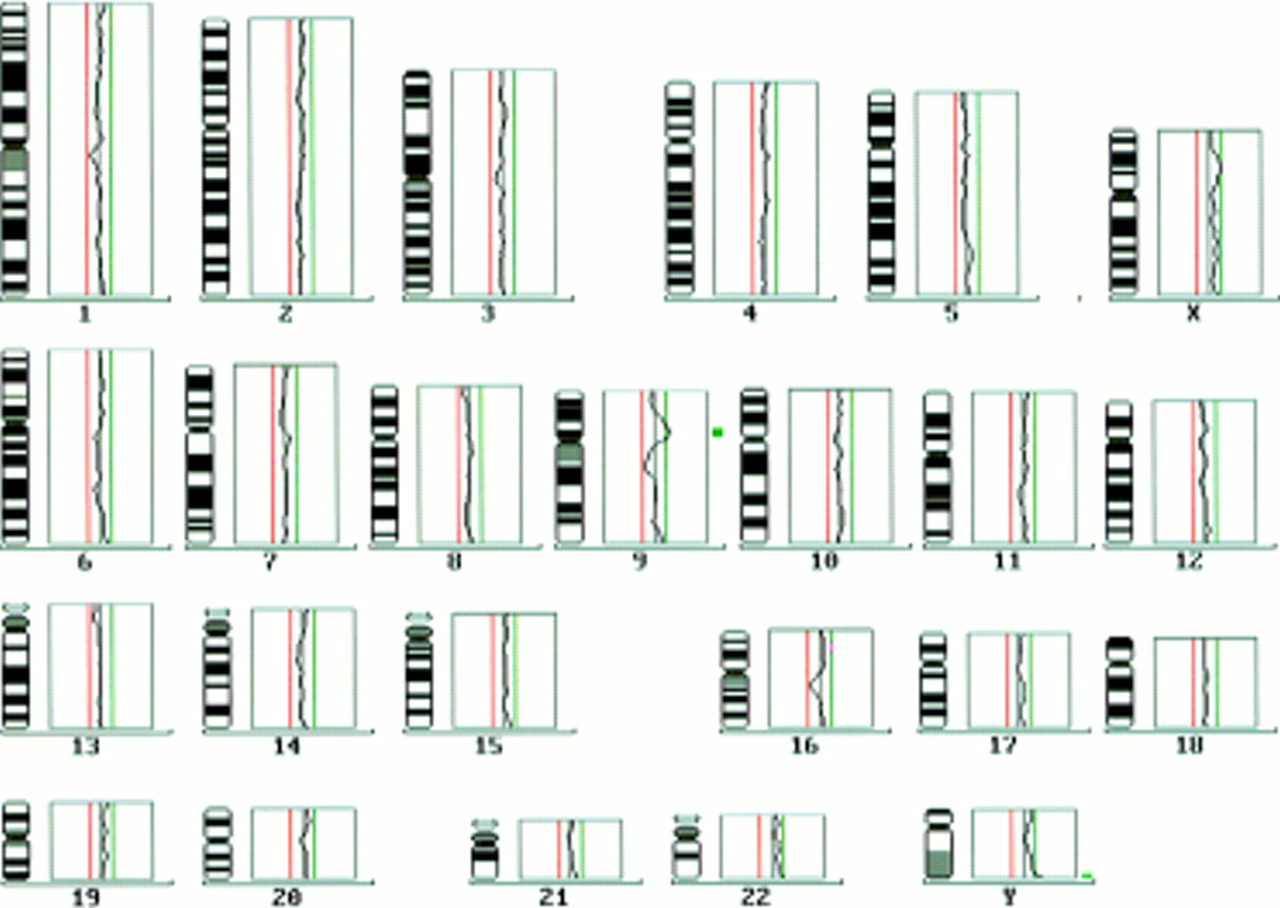
Comparative genomic hybridisation (CGH) was applied to identify the additional material inserted in the short arm of chromosome 5. CGH showed a gain of material in the proximal region of the short arm of chromosome 9 (rev ish enh(9)(p12p21)) (fig 3). A further WCP using a chromosome 9 specific probe confirmed the CGH result and detected an insertion of chromosome 9 material in the short arm of chromosome 5 (fig 4).

CGH profiles of the proband's (II.2) chromosomes. The thin vertical lines represent ration profile value of 0.5 and 1.5. The average profile of chromosome 9 shows amplification of proximal 9p.

Partial metaphase of II.2 showing whole chromosome painting with a chromosome 9 specific probe. A chromosome 9 specific signal is visible in the short arm of chromosome 5.
mBand and WCP analysis on metaphases of the cri du chat syndrome patient (III.1) with chromosome 5 and chromosome 9 specific probes showed that the breakpoint of the deleted chromosome 5 occurred in the inserted region of the paternally inherited derivative chromosome 5. The chromosome 5 breakpoint could be determined by mBand at 5p13.3 (fig 5). Therefore, patient III.1 has a partial monosomy (5)(p14pter) and a partial trisomy 9p. The origin of the der(5) telomere remains unclear.
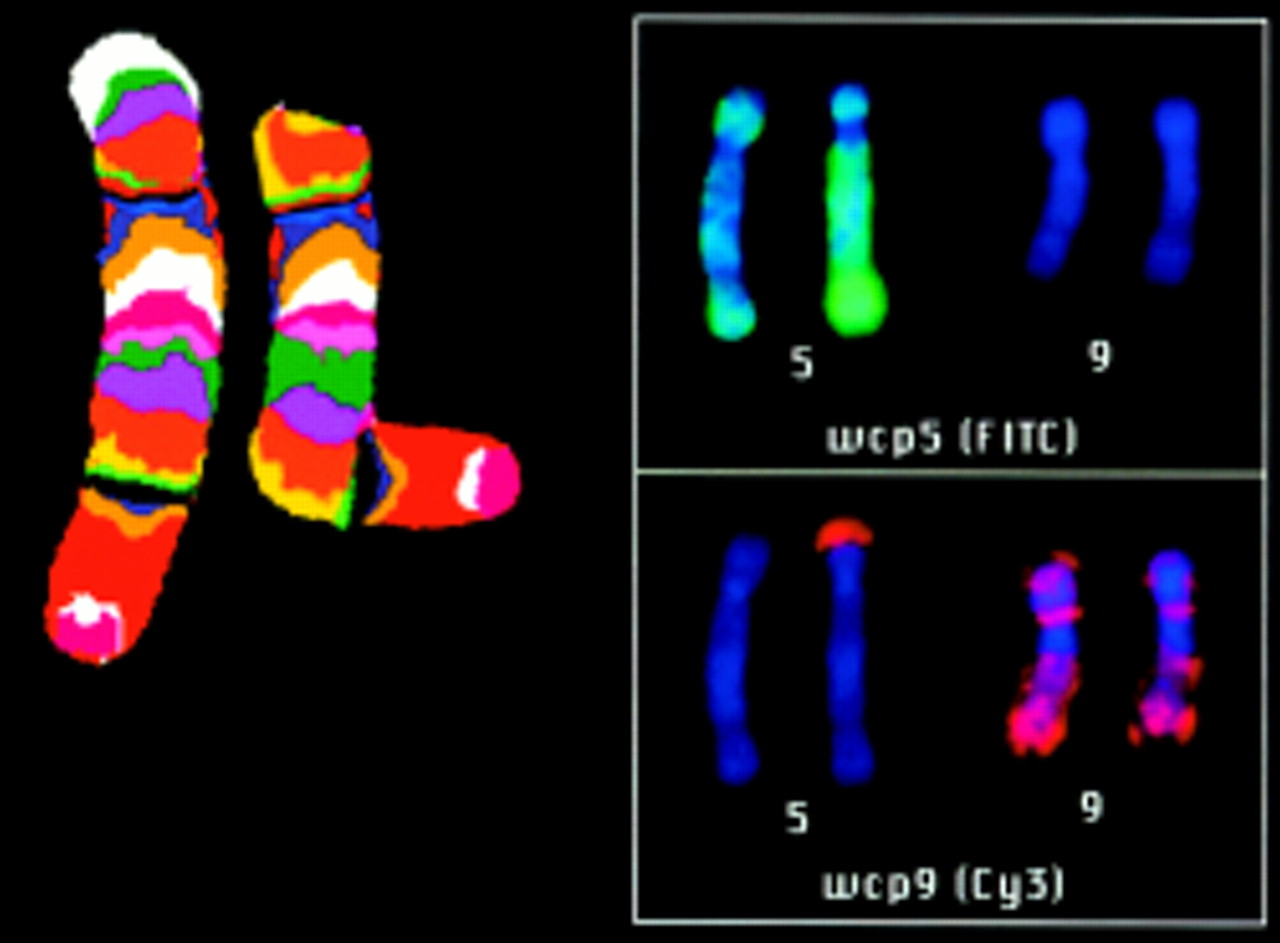
Chromosome 5 mBand-FISH determined the chromosome 5 insertion breakpoint in III.1 at 5p13.4 WCP 5 and WCP 9 showed the breakpoint of the 5p deletion in the inserted region.
Chromosome analyses of the mother (I.2) and the brother (II.3) of II.2 showed a balanced insertion of material from the short arm of chromosome 9 into the short arm of chromosome 5 (ins(5;9)(p13.3;p12p21)). The GTG banding results were confirmed by FISH using chromosome 5 and chromosome 9 specific whole chromosome painting probes (fig 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Metaphase of I.2 showing a whole chromosome painting with specific probes for chromosomes 5 (red) and 9 (green). A shortened chromosome 9 specific signal is visible on der(9). The missing segment is inserted in the short arm of the der(5).
For a better characterisation of the trisomic region, microsatellite analysis with seven markers from chromosome 9p and one marker from chromosome 9q was performed on DNA from I.2, II.2, II.3, and III.1. Most markers in the critical region were not informative. Only microsatellite marker D9S104 showed three different alleles in III.1, confirming a trisomy for the subband 9p21 (data not shown). Therefore, YAC-FISH was performed to determine the trisomic region in more detail. According to the GTG banding and CGH results, the trisomic segment was postulated to include the chromosomal subbands 9p12-9p21.3. Five different YAC probes localised within and two YAC probes localised outside this region were hybridised to metaphases of II.2 (table 1). All YACs from the critical region 9p12-p21.3 resulted in three specific signals, two on the normal chromosomes 9 and one on the derivative chromosome 5. In contrast, the YACs which are localised in 9p23 and 9p24 only showed signals on both chromosomes 9.
Results of the YAC-FISH with probes spanning the chromosomal region 9p12 to 9p24
Taking all the cytogenetic results together, II.2 is a carrier of an unbalanced maternally inherited inserted translocation (5;9)(p13.3;p12p21.3) resulting in a partial trisomy 9p12-p21.3. This case shows the efficient use of CGH, WCP, and mBand for the correct identification of chromosomal material of unknown origin and of the mechanisms generating structural chromosome aberrations.
DISCUSSION
Chromosomal insertions, with an estimated frequency of less than 1 in 5000 newborns, are relatively rare chromosome rearrangements.7 Only a few reports of inherited interchromosomal insertions have been published and most cases have arisen de novo. Insertions may lead to disruption of genes, position effects with alterations of gene function, or loss or acquisition of chromosomal segments that cannot be detected by cytogenetic methods. Unbalanced transmission of insertion chromosomes results in partial trisomies and/or monosomies. We report on an unbalanced inherited maternal insertion (5;9)(p13.3;p12p21.3) resulting in partial trisomy 9p12-p21.3. To the best of our knowledge, this is the first report of partial trisomy 9p and a normal phenotype. The normal phenotype is unexpected, because the unbalanced insertion includes a trisomic segment of about 21 Mb (Entrez Map View Chromosome 9, Genes on Sequence Map), harbouring about 280 genes (Entrez Map View, Chromosome 9, Genes on Cytogenetic Map). It is usual that such large autosomal trisomies result in mental retardation and morphological anomalies. In particular, partial trisomy 9p is a well described phenotype and results in a complex clinical malformation pattern. Haddad et al8 reviewed the phenotypic effects of different trisomic regions of chromosome 9p. They compared the cytogenetic and clinical findings of 144 previously published cases with partial or complete trisomy 9p. The majority of these cases were caused by an unbalanced translocation involving another chromosome. Phenotypic effects of these additional segments could not be excluded in these cases. Therefore, the authors confined their analysis to 11 patients with direct duplications of 9p. There was a remarkable consistency in the facial and digital anomalies, which were present in all patients. A general trend towards a milder phenotype in cases with smaller and more distal duplications could be suspected. Furthermore, the characteristic trisomy 9p phenotype showed the best correlation with the 9p22 region. FISH analyses by Fujimoto et al9 and Guanciali Franchi et al10 suggest that the segment 9p22 may be the critical region for the duplication 9p syndrome. Band 9p22 was shown to be duplicated in all patients with de novo duplications of 9p without involvement of another chromosome abnormality. Just one patient11 showed a duplication without involvement of 9p22. Very limited information is available on this patient and the precise breakpoint must be questioned because chromosome analysis was performed at a low banding level.9 These findings could also explain why our proband showed no phenotypic abnormalities, because the “critical” region 9p22-9p24 does not seem to be involved in the partial trisomy 9. However, the normal phenotype in II.2 suggests that the genes located in the trisomic segment may not be subject to dosage effect.
A further case with a combination of cri du chat syndrome and partial trisomy 9p was described by Sigmund et al.12 In this case, the combination resulted from an unbalanced translocation (5;9)(p13.3;p13.1). The patient harboured the “critical” segment 9p22 and showed typical clinical features of both chromosomal abnormalities.
Imprinting effects may also be an explanation for the unexpected phenotype in our patient. However, clear imprinted regions on chromosome 9 have not been confirmed so far (Imprinted Gene Catalogue Records, 2000).
It is difficult to estimate the reproductive risk for further children of II.2, because he has an interchromosomal duplication resulting from an unbalanced inherited maternal insertion. Insertions can be associated with a very high reproductive risk. The average risk of insertion carriers to have a child with an abnormal phenotype is in the range of 10-50%. Two possibilities of segregation in meiosis have to be considered in our case. Firstly, the risk for the transmission of the rearranged chromosome 5 in II.2 is a priori 50%, which is higher than the risk for a straightforward insertion. However, the risk for an abnormal phenotype must be lower, because II.2 has a normal phenotype. Secondly, there is also a risk for a deletion in 5p, which can be best explained by a synapsing of the homologous chromosomes 5 in meiosis I, followed by a looping out of the insertional segment and a break inside the loop.7 However, this deletion risk is difficult to estimate and no specific published data are available. All in all, a detailed risk estimation cannot be given in the present case, but a prenatal diagnosis should be performed in further pregnancies of II.2, as well as of his brother.
For further analysis of the unexpected genotype-phenotype correlation of patient II.2, we have established a lymphoblastoid cell line of this patient. Expression analysis by array technologies may show whether the trisomic segment alters the gene expression profile in these cells.
Acknowledgments
The authors thank Mrs R Drange, Mrs B Bütow, and Mrs B Plückthun for excellent technical assistance. We also thank Dr J Wirth from the Molecular Cytogenetic and Positional Cloning Centre of the MPI of Molecular Genetics in Berlin for providing YAC-DNA and Mrs K Saar from the Microsatellite Centre of the Max-Delbrück-Zentrum in Berlin for performing microsatellite analysis. Furthermore, we thank Metasystems for paying for the colour figures.
Electronic database information. Entrez Genome: http://www.ncbi.nlm.nih.gov/Entrez/ Imprinted Gene Catalogue Records: http://cancer.otago.ac.nz/IGC/Web/home.html The Molecular Cytogenetic and Positional Cloning Center: http://www.molgen.mpg.de