Article Text

Abstract
Background and objective: In clinical settings with fixed resources allocated to predictive genetic testing for high-risk cancer predisposition genes, optimal strategies for mutation screening programmes are critically important. These depend on the mutation spectrum found in the population under consideration and the frequency of mutations detected as a function of the personal and family history of cancer, which are both affected by the presence of founder mutations and demographic characteristics of the underlying population. The results of multistep genetic testing for mutations in BRCA1 or BRCA2 in a large series of families with breast cancer in the French-Canadian population of Quebec, Canada are reported.
Methods: A total of 256 high-risk families were ascertained from regional familial cancer clinics throughout the province of Quebec. Initially, families were tested for a panel of specific mutations known to occur in this population. Families in which no mutation was identified were then comprehensively tested. Three algorithms to predict the presence of mutations were evaluated, including the prevalence tables provided by Myriad Genetics Laboratories, the Manchester Scoring System and a logistic regression approach based on the data from this study.
Results: 8 of the 15 distinct mutations found in 62 BRCA1/BRCA2-positive families had never been previously reported in this population, whereas 82% carried 1 of the 4 mutations currently observed in ⩾2 families. In the subset of 191 families in which at least 1 affected individual was tested, 29% carried a mutation. Of these 27 BRCA1-positive and 29 BRCA2-positive families, 48 (86%) were found to harbour a mutation detected by the initial test. Among the remaining 143 inconclusive families, all 8 families found to have a mutation after complete sequencing had Manchester Scores ⩾18. The logistic regression and Manchester Scores provided equal predictive power, and both were significantly better than the Myriad Genetics Laboratories prevalence tables (p<0.001). A threshold of Manchester Score ⩾18 provided an overall sensitivity of 86% and a specificity of 82%, with a positive predictive value of 66% in this population.
Conclusion: In this population, a testing strategy with an initial test using a panel of reported recurrent mutations, followed by full sequencing in families with Manchester Scores ⩾18, represents an efficient test in terms of overall cost and sensitivity.
- MGL, Myriad Genetics Laboratories
- NCI, National Cancer Institute
- PCR, polymerase chain reaction
- ROC, receiver operating characteristic
Statistics from Altmetric.com
- MGL, Myriad Genetics Laboratories
- NCI, National Cancer Institute
- PCR, polymerase chain reaction
- ROC, receiver operating characteristic
Breast cancer is now the most common cancer and the leading cause of death from cancer in women worldwide.1 Information on family history is key to assessing the clinical relevance of gene variants to help refine risk prediction and to target interventions at high-risk individuals who have the most health benefits to gain from available preventive and risk reduction strategies.2–6 Like other common cancers, breast cancer shows some degree of familial clustering, with the disease being 2–4-fold more prevalent in first-degree relatives of affected people,7–9 and twin studies suggest that most of the excess familial risk results from inherited susceptibility.10,11BRCA1 (MIM 113705) and BRCA2 (MIM 600185) are the most important breast cancer susceptibility genes identified to date, being responsible for about 20–25% of the excess risk, and for a much larger proportion of hereditary breast or ovarian cancer occurring in families with multiple cases of cancer.12–16 Predictive genetic testing for these high-risk cancer predisposition genes is increasingly part of clinical practice.6,17,18 In settings with fixed resources allocated to such testing, such as in countries with nationalised healthcare, optimal strategies for cost-effective mutation screening programmes are critically important.
The prevalence of BRCA1 and BRCA2 germline mutation carriers in families with breast and/or ovarian cancer depends on the number, type of cancer and age at diagnosis of affected individuals, as well as the demographic characteristics of the underlying population studied. Founder mutations have been identified in numerous populations as well as across populations.19 For genetic testing, there are several advantages of knowing the characteristics of recurrent mutations in a founder population. For instance, a more accurate estimate of the prior probability of carrying a mutation should be possible, and testing can be targeted to the founder or recurrent mutations, allowing a more rapid and less expensive initial test, reaching an adequate sensitivity and specificity. Evidence for such founder mutations in the French-Canadian population was reported soon after the identification of the BRCA1 and BRCA2 genes, as well as during the progress of this study.13,20–25
Estimations of age-specific risks for breast and ovarian cancer vary by birth cohort, mutation characteristics, ascertainment criteria, population studied and the family history of the disease of the index case.16 For instance, population-based studies estimated the risk of breast cancer by age 70 years in BRCA1 and BRCA2 carriers to be 65% and 45%, respectively, whereas the corresponding risk of ovarian cancer was 39% and 9%, thus being less than that estimated in families with multiple cases of cancer.15,26 In addition, estimates derived from other populations cannot necessarily be extrapolated to founder populations, such as the French-Canadian,27,28 and these risk estimates are important for genetic counselling.29
To maximise the available resources for often expensive genetic analysis of the BRCA1/BRCA2 genes, several investigators have developed predictive algorithms for testing on the basis of personal or family history to select families that are most likely to carry a deleterious mutation in BRCA1 or BRCA2.30 These include several methods based on specific genetic models using complete pedigree information, such as BRCAPRO31 and Breast and Ovarian Analysis of Disease Incidence and Carrier Estimation Algorithm,32 as well as a number of more empirical approaches based on family history.30 The relative merits of the two genetic model approaches in a subset of this cohort have been recently assessed29; therefore, in this study, we have examined two of the more commonly used empirical approaches. The first is the so-called Manchester Scoring System that was developed to estimate the probability of identifying mutations in BRCA1 and BRCA2 genes.33,34 Using a combination of results from screening and the family history of kindreds with and without mutations, this simple scoring system was devised to predict pathogenic mutations and particularly to discriminate at the 10% likelihood level. An initial cut-off at 10 points was thought to equate to >10% probability of a deleterious mutation in BRCA1 and BRCA2, but this was recently updated in a larger, more completely screened set, and a score of ⩾15 was proposed for the 10% mutation probability. The second approach is based on the large set of personal and family history data collected by Myriad Genetics Laboratories (MGL), Salt Lake City, Utah, USA, in which a table stratifying the personal cancer history of the probands by their family history of breast and ovarian cancer is produced, with each entry in the table reporting the prevalence of deleterious mutations found among all individuals with that particular combination of family and personal history.35,36
The operating characteristics, overall utility and choice of approach clearly depends on both the specific population and the particular clinical setting to which the algorithm is to be applied, as both affect the prior probability of finding a mutation, and hence the predictive value of the test. Thus, it is important to examine these issues in a wide range of populations and clinical settings. In this regard, the two empirical approaches described above have also been compared with a logistic regression approach based on the data from this study.
To deal with these and other questions in familial breast and ovarian cancer in Quebec, we undertook a large multidisciplinary project in the province of Quebec, with family collection, laboratory, clinical, epidemiological, psychosocial, ethical, legal, and social issues components. This translational clinical research programme is part of the ongoing INHERIT BRCAs (INterdisciplinary HEalth Research International Team on Breast CAncer susceptibility) Study. The major goals of this interdisciplinary team approach have been published previously.37,38
The aims of this study were (1) to examine the contribution of both founder/recurrent and novel rare mutations to multiple cases of breast and ovarian cancer in families in Quebec and to evaluate the geographical distribution of these alleles in the population; and (2) to devise an optimal strategy for clinical genetic testing of French-Canadian families with breast and ovarian cancer, incorporating both the molecular approach and family selection criteria. In particular, given the typically large structure of French-Canadian families, we wished to devise the most relevant definition of “family” to be used in the categorisation of family history. This definition will directly improve the clinical validity of such predictive tests.
METHODS
Ascertainment of families
The recruitment of French-Canadian families with high risk of breast or ovarian cancer started in 1996 through a research project, which thereafter evolved into a large ongoing interdisciplinary research programme designated INHERIT BRCAs.37–39 The recruitment of index cases was made possible by a network of referring clinicians across the province of Quebec. Potential participants were first approached by their clinicians who explained to them the nature and objectives of the research project. Thereafter, individuals interested in the project sent a signed form authorising a member of the research team to contact them. A total of 571 index cases have been referred to our study. After this form had been received, an initial telephone contact was made to explain briefly the procedure of the research project, to evaluate the eligibility of the family and to make an appointment for a pretest education session. Family history was recorded, and the pedigree analysed. Index cases were recruited if their family met one of the following strict criteria: (1) at least four individuals with breast or ovarian cancer diagnosed in first-degree or second-degree relatives; and (2) three individuals with breast or ovarian cancer in first-degree relatives.
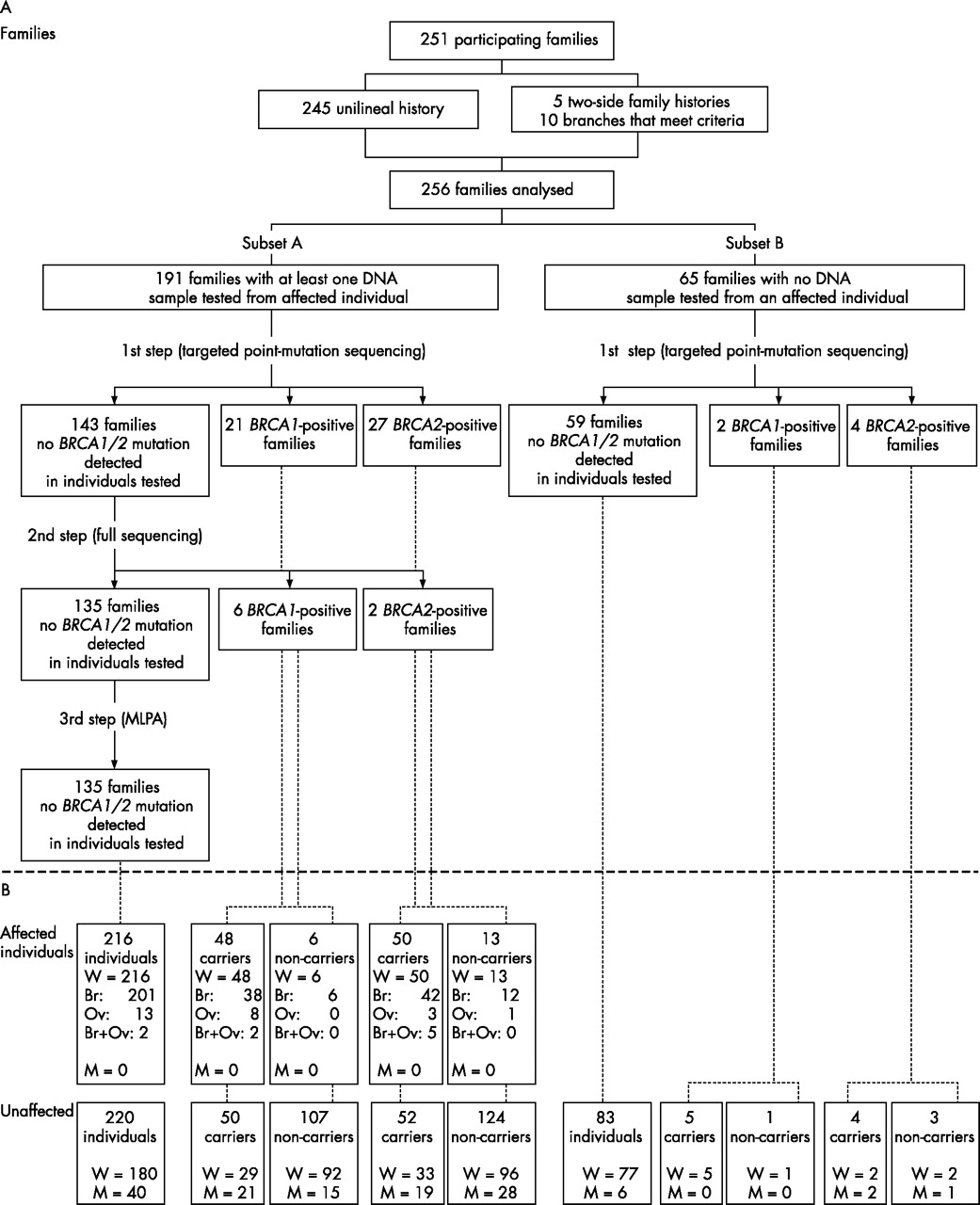
Of the 571 index cases, 399 had family histories satisfying these criteria; 148 index cases decided not to undergo genetic testing or were found to be ineligible after receipt of pathology records, leaving a total of 251 participating families in this study. Moreover, as five index cases had a strong history of breast and/or ovarian cancer on both the paternal and maternal sides, two separate branches were created for each of these families and another index case was designated in the branch with the weaker history. Thus, the analyses presented in this study were performed on 256 families, which have been divided into two groups: subset A consisting of 191 families for which at least one DNA sample from a woman with breast or ovarian cancer was available for genetic testing, and subset B consisting of 65 families for which no DNA sample from an affected woman was available. Figure 1 represents the overall ascertainment scheme.

(A) Classification of participating families based on the availability of a DNA sample from an individual with breast (Br) and/or ovarian (Ov) cancer and the BRCA1/BRCA2 mutation status. (B) Distribution of affected and unaffected individuals. M, men; MLPA, multiple ligation-dependent amplification; W, women.
Index cases deciding to participate signed a consent form, and a blood sample was then taken. Relatives were recruited by the index case who was invited to distribute a form authorising a member of the research team to contact them. All participants had to be at least 18 years old and mentally capable. Finally, participants consenting to be informed of their results were seen by a clinician from one of the seven participating hospitals, who was directly involved in this translational research programme and responsible for the disclosure of genetic test results. The result disclosure session included a review of medical recommendations pertaining to each participant’s test result, and a discussion of the possible individual and familial effects of their result. Pathology reports were requested to first confirm all diagnoses of breast and ovarian cancer, followed by BRCA-related cancers such as those of the pancreas, prostate, colon and larynx. When available, other cancers were also confirmed. We tried to have confirmation for all affected participants and for all first-degree relatives of a study participant; about 95% of breast or ovarian cancers were confirmed in participants. The study was approved by the appropriate ethics committee at each participating institution.
DNA extraction, primer design, polymerase chain reaction amplification and sequencing reactions
After signed informed consent had been obtained from each participant, 40 ml of blood was drawn. The blood was separated into two coded tubes, and genomic DNA was extracted independently by two technicians using standards methods at the Cancer Genomics Laboratory, Quebec City, Quebec, Canada. Confirmation tests were performed on a second blood sample for each individual belonging to a family positive for BRCA1 or BRCA2. One tube was used for an independent confirmation performed at the Cancer Genomics Laboratory, and another tube was sent to the Molecular Diagnostic Laboratory, Alberta Children’s Hospital, Calgary, Alberta, Canada, for confirmation of results (carrier or non-carrier of a known familial mutation).
Each region in which a mutation of interest was located was sequenced twice using different sets of polymerase chain reaction (PCR) and nested sequencing primers on the two different genomic DNA samples extracted from each individual. The PCR and sequencing primers used for amplifying genomic DNA for testing each specific mutation are given in the table available online at http://jmg.bmj.com/supplemental. Primers were carefully designed to avoid known single-nucleotide polymorphisms reported in the Breast Information Core database (http://research.nhgri.nih.gov/bic/). PCRs were amplified on a GeneAmp 9700 thermocycler (Applied Biosystems, Foster City, California, USA) using standard methods. After purification of PCR products with silica unifilter microplates (Whatman, Florham Park, New Jersey, USA), the concentration was determined by fluorescence (Picogreen dye, Invitrogen Canada, Burlington, Ontario, Canada) using a FL600 Microplate Fluorescence Reader (Bio-Tek Instruments, Highland Park Winoski, Vermont, USA). The sequencing reactions were purified using the isopropanol precipitation or ethanol–EDTA procedure as described by the manufacturer (Applied Biosystems). Finally, all reactions were migrated through an ABI-3700 or ABI-3730xl automated sequencer (Applied Biosystems). Sequence analyses were performed using the Unix Staden package (Cambridge University, Cambridge, UK).
A list of PCR and sequencing primers used for sequencing unclassified variants is available on request. The primers and conditions used by the Molecular Diagnostic Laboratory, Alberta Children’s Hospital, to confirm our results are also available on request.
Multistep mutation testing
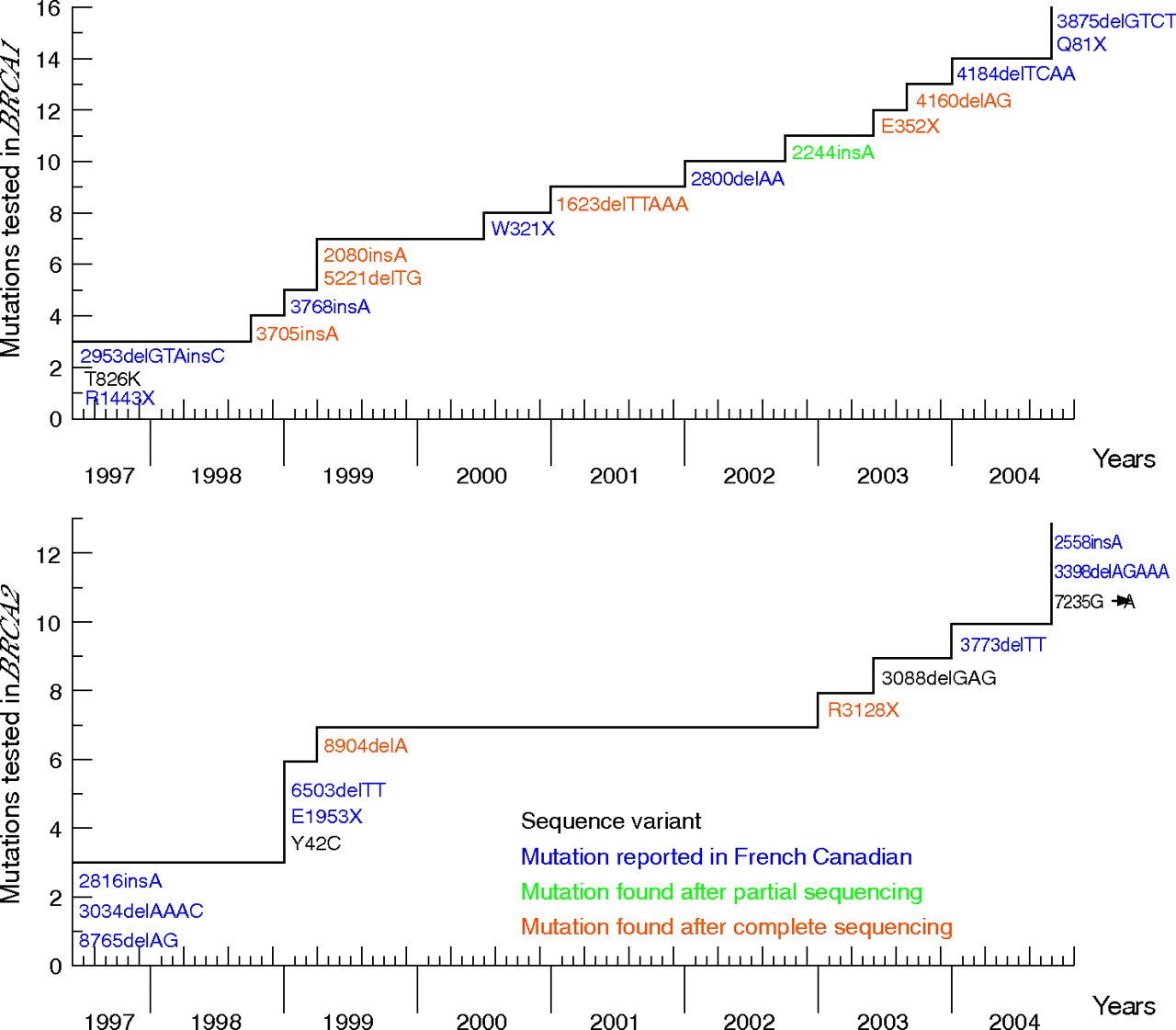
As the number of detected or reported mutations in the French-Canadian population has increased during the course of this study, participants were first tested for the panel of known mutations at the time of their entry into the study. Figure 2 depicts the evolution of the mutation testing during the course of this study. Each time a new deleterious mutation was published in the French-Canadian population, or was found after targeted point mutation testing in this study or by a full sequence analysis (MGL) performed before December 2004, it was included in the panel of mutations tested (fig 2). A few unclassified sequence variants that initially seemed to have a potential effect on BRCA1 or BRCA2 proteins (T826K in BRCA1, Y42C in BRCA2), or for which cosegregation with the disease in the family could not be excluded (3088delGAG in BRCA2), were also included. To date, each participant’s genomic DNA sample has been tested for a panel of 29 mutations (19 frameshift, 6 nonsense, 2 missense variants, 1 splice and 1 in-frame deletion). For each family that received a non-conclusive result for BRCA1 or BRCA2 (ie, no mutation was found by screening for the panel of mutations analysed), at least one DNA sample from a woman with breast or ovarian cancer (with the exception of four DNA samples from an unaffected parent from whom the familial history of breast/ovarian cancer was derived) was sent to MGL for full-length BRCA1/BRCA2 sequencing following their Comprehensive BRACAnalysis-BRCA1 and BRCA2 gene sequence analysis for susceptibility to breast and ovarian cancer test. Testing services were performed according to the Memorandum of Understanding of the National Cancer Institute (NCI) for NCI-funded research testing services for BRCA1 and BRCA2 (project number NCI 173). A DNA sample from the youngest recruited patient with breast or ovarian cancer was systematically sent for full-length BRCA1/BRCA2 sequencing. Finally, DNA samples from all 135 families in which no deleterious mutation was found were tested at the Cancer Genomics Laboratory by multiplex ligation probe amplification for both BRCA1 and BRCA2 genes. As recently described, this approach failed to detect any deleterious rearrangement in this population.40

Schematic representation of the evolution of BRCA1 and BRCA2 mutation testing over the course of this study. Deleterious mutations in the French-Canadian population reported in the literature were 2953delGTAinsC,22 R1443X,21 3768insA,24 W321X (Dr Elizabeth Springs, personal communication, 2000) and 2800delAA,41 4184delTCAA (Breast Cancer Information Core, http://research.nhgri.nih.gov/bic/), 3875delGTCT and Q81X25 in the BRCA1 gene; 2816insA (BCLC meeting, Lyon 1996, unpublished data,24 3034delAAAC,13 8765delAG,23 6503delTT and E1953X,24 3773delTT (Breast Cancer Information Core, http://research.nhgri.nih.gov/bic) and 2558insA, 3398delAGAAA and 7235G→A25 in the BRCA2 gene; and the sequence variant, T826K, in which a potential deleterious effect was suspected in the BRCA1 gene.20
Genealogical reconstruction and analyses
The ascending genealogy of the 246 participating families who gave their consent to this component of the project was reconstructed. From each family harbouring a BRCA1 or BRCA2 mutation, one carrier individual was chosen as the genealogical proband. Among the remaining families, one affected individual was selected. In most instances (96%), the genealogical proband was the index case, a parent or a member of his or her sibship. In the remaining 4% of cases, the affected individual was a close relative. All genealogies were reconstructed as far back as sources allowed and, in most cases, lineages were traced back to the first immigrants to Canada. Genealogical reconstruction relied on the BALSAC population register and on the BALSAC-RETRO database which contains linked genealogical information on nearly 375 000 individuals married in Quebec between the beginning of the 17th century and 2005.42 When necessary, complementary sources such as the Population Register of Early Quebec (Programme de Recherche en Démographie Historique: http://www.genealogie.umontreal.ca/en/), genealogical dictionaries, marriage registries or microfilms of parish registers were also consulted. In four instances, reconstructions were unsuccessful because of a lack of information or adoptions. Therefore, a total of 242 genealogies were analysed.
To describe the general characteristics of the genealogical data, some parameters, such as the completeness index, which were measured, gives, for each generation level, the proportion of ancestors who have been successfully identified. This index is also used to calculate the mean generation level at which the lineages in the genealogies come to an end (average genealogical depth). Places of marriage of probands’ grandparents were obtained from the BALSAC database, and their distribution in Quebec was mapped. This could provide clues to the diffusion patterns and current geographical distribution of BRCA1/BRCA2 mutations in Quebec. In a previous study, we have detected such indications of spatial concentration of the BRCA1 R1443X mutation.43
Statistical analysis
Multiple stepwise logistic regression analysis was used to evaluate the importance of measures related to family history in predicting BRCA1 and BRCA2 carriers. These analyses included only the 191 families in subset A in which an affected individual had been screened for mutations. In all, 27 of the families had deleterious BRCA1 mutations, 29 had BRCA2 mutations and the remaining 135 had no mutation after complete direct sequencing and a test for large genomic deletions or rearrangements using multiple ligation-dependent amplification analyses.40 Factors examined were the presence of male patients with breast cancer in the family (yes/no); number of breast cancers diagnosed at ages <50 and ⩾50 years (coded as 0–1, 2–3, ⩾4); and the number of cases of ovarian cancer in the family without regard to age (0, 1, ⩾2). In counting the affected relatives, only women with breast cancer and ovarian cancer who were third-degree relatives or closer to the index case and men with breast cancer at any degree from the index case were considered. Separate stepwise logistic regression analyses comparing BRCA1 with no mutation detected and BRCA2 with no mutation detected were performed, and variables not significantly predictive at a level of 0.001 were eliminated. The final retained models are presented in table 4. To use the logistic model to estimate the probability that a given family would have a BRCA1/BRCA2 mutation, we used a Jacknife procedure in which the above-mentioned logistic regression model was re-estimated leaving each family out of the estimation in turn, and then using the estimated parameters to estimate the probability of that family having a mutation. Thus, the parameters used in the prediction of each family are not derived from that family’s data.
The mean age at onset of breast or ovarian cancer in women was compared among the three groups of families using analysis of variance, with the mean age at diagnosis for each family being weighted by the number of cases in that family. For each case where the overall effect of family group (BRCA1+ vBRCA2+ v no mutation detected) was significant, we performed two retrospective comparisons: BRCA1+ versus BRCA2+ and BRCA1/BRCA2+ versus no mutation detected.
To test the utility of two simple scoring systems in predicting the BRCA mutation status, we first used the Manchester Scoring System developed by Evans et al.33,34 Specifically, we calculated both the BRCA1-associated and BRCA2-associated Manchester Scores for both the maternal and paternal lineages of the index case. The maximum of these four scores was then taken as the Manchester Score for the family and used in subsequent analyses. In addition, we obtained the predicted probability of a BRCA1/BRCA2 mutation based on the personal history of the tested individual and the cancer history in relatives using the prevalence tables obtained at http://www.myriadtests.com/provider/mutprev, last updated in June 2005, and based on results of full sequence testing of >37 000 non-Ashkenazi individuals.
Using these scores, receiver operating characteristic (ROC) curves were constructed, showing sensitivity plotted against 1−specificity. Here, sensitivity is defined as the proportion of families with a BRCA1/BRCA2 mutation with a score or probability greater than or equal to a given threshold plotted against the proportion of families with scores below the threshold without an identified BRCA1/BRCA2 mutation. One point on the curve is obtained for each possible threshold. The area under the curve is used as a measure of the adequacy of the criteria used for genetic testing. The two models providing a predicted probability of having a mutation (logistic regression and Myriad prevalence table) were evaluated in terms of the Brier Score.44 The smaller the Brier Score, the closer the predictions to the observed data; the Spiegelhalter z statistic provided a significance test of the adequacy of the fit.
All statistical analyses were performed using STATA V.8.0.
RESULTS
Descriptive parameters of the cohort study
A total of 982 participants were recruited from 256 high-risk families with breast or ovarian cancer (fig 1). With the exception of one man, all index cases were women, of whom 31% had breast or ovarian cancer at their entry into the study. Participants were predominantly women (86.5%), without breast or ovarian cancer (63.4%), and about 75% of them were <60 years old (more prevalent in their 40s (26.7%) and 50s (28.2%); table 1). In all, 79.2% of participating women were parous and 63.1% were post-menopausal at the time of entry into the study.
Participants’ characteristics
In the 242 genealogies analysed, >85% of the ancestors were traced up to the ninth generation; from then on, more and more ancestors were immigrants, and completeness dropped rapidly to 50% at the 11th generation and to 3% at the 13th generation. The maximum depth was 17 generations and was observed in a few lineages. The genealogies had an average depth of 10 generations. Only 8 families had a mean genealogical depth <8 generations, with the lowest value at 5.2 generations. This corresponds to >240 years of mostly French-Canadian ancestry for >95% of participating families based on an average intergenerational interval of 30 years.45
Mutation testing outcome
Targeted point-mutation sequencing of observed or published mutations found in French-Canadian families (fig 2) constituted the first screening step of genetic analysis of BRCA1 and BRCA2 mutations in our cohort. This approach led to the identification of seven deleterious mutations (2244insA, 2953delGTAinsC and R1443X in BRCA1; 2816insA, 3034delAAAC, 6503delTT and 8765delAG in BRCA2) in 54 different families (table 2).46–53 The mutation 2244insA was the only deleterious mutation found after such a targeted sequencing of a BRCA1 region containing another mutation. To date, this mutation has not been reported in the Breast Cancer Information Core database.
Deleterious mutations and sequence variants identified in French-Canadian families at high risk of breast or ovarian cancer
The second step consisted of a full sequence analysis of BRCA1 and BRCA2 genes in each family not found to have one of the above specific mutations and for which a DNA sample from an affected individual (or obligate carrier) was available for testing (subset A), as described in the Methods section. This second-step screening approach led to the discovery of eight additional deleterious mutations (E352X, 1623delTTAAA, 2080insA, 3705insA, 4160delAG and 5221delTG in BRCA1; 8904delA and R3128X in BRCA2), each found in a different family (table 2).
By using this two-stage approach, a deleterious mutation was identified in 62 of the 256 (24%) families (29 in BRCA1, 33 in BRCA2). Of these, 56 were found in the 191 (29%) families of subset A. Among these 27 BRCA1-positive and 29 BRCA2-positive families, 48 (86%) were found to harbour a mutation detected by the first step targeted point sequencing. On the other hand, 6 of 65 (9%) families from subset B were BRCA1/BRCA2 positive. Owing to the lack of available DNA samples from affected individuals from subset B, complete BRCA1/BRCA2 sequencing was not performed in any families from this subset of 65 families, and mutation-negative status is therefore based on only the first-step screening approach. Figure 1 also shows that in the 62 positive families, 209 carriers were identified, including 98 affected women, 69 unaffected women and 42 unaffected men, whereas 254 individuals were non-carriers, including 210 women, among whom 19 were affected.
Nine BRCA1 and six BRCA2 distinct deleterious mutations were detected in our cohort (table 2). Eleven mutations were found to occur in only one family, whereas four recurrent mutations were identified—namely, 2244insA (n = 3), 2953delGTAinsC (n = 2) and R1443X (n = 18) in BRCA1 and 8765delAG in BRCA2 (n = 28; table 2). These four mutations can explain 82% of mutation-positive families from subset A and 83% of mutation-positive families from subset B. In subset A, the two most frequent mutations (R1443X and 8765delAG) correspond to 75% of BRCA1/BRCA2-positive families (table 2).
Other sequence variants identified in our cohort
The full sequence analyses performed in this study led to the detection of 27 sequence variants (table 2). Their classification was primarily based on MGL observations and on several published studies.46–53 Eight of them have been classified, during the course of our study, as “favour polymorphism”: S741F, P1099L and R1347G in BRCA1; Y42C, D1420Y, V2728I, K3326X and the read-through variant 10481delTCTA in BRCA2 (table 2). Further, sequence variants N417S in BRCA1, and E462G and R2034C in BRCA2 were classified as having neutral/little clinical significance. Finally, 16 sequence variants still remain unclassified or with an uncertain clinical significance. These were considered to be mutation-negative for the purpose of this study. Table 2 also shows that variants Y42C, E462G, 3088delGAG and Q2858R were found to co-occur with a deleterious mutation in five French-Canadian BRCA1/BRCA2-positive families.
Regional distribution of deleterious BRCA1/BRCA2 alleles
Families at high risk of breast or ovarian cancer were recruited from all regions of Quebec as shown on the map of the regional distribution of index cases’ places of residence (fig 3A). The greatest number of families is found in the area of Quebec City (42%) and in the eastern part (32%) of the province, where most doctors collaborating with our study are based. Three families were from New Brunswick, but were recruited through referring centres in Quebec. To minimise the effect of inter-regional migrations (mostly from rural to urban areas) that took place in the past few decades and to obtain some insight into the regions of origin of the mutations, we also mapped the place of marriage of genealogical probands’ grandparents (fig 3B). Nineteen pairs of grandparents were married in the US, Ontario or in a place that could not be identified, and they are not represented on the map. Both maps show the proportion of the two most common founder mutations, all other deleterious mutations and families with no mutation identified in each region. The proportion of families in which a mutation was identified varies considerably according to region, and this is more conspicuous when looking at the region of origin of probands as shown by the distribution of grandparents’ place of marriage (fig 3B). A mutation was identified in 24.7% of all families, but fig 3B indicates that in the regions of Bas-St-Laurent (01), Mauricie (04) and Lanaudière (14), this proportion lies between 40% and 50%. However, these figures should be interpreted with caution, as in some regions the number of recruited families is quite small, as shown by the size of the circles.

(A) Place of residence of index cases in the Quebec territory (n = 251) and (B) place of marriage of genealogical probands’ grandparents (n = 465). The size of the circles is proportional to the number of high-risk families recruited in each of the regions. Parts in the pies represent, for each region, the proportion of families harbouring one of the two founder mutations (BRCA1, orange; BRCA2, blue), another BRCA mutation (yellow) or for whom testing was inconclusive (red). Numbers on the map represent the following regions: (01) Bas St-Laurent, (02) Saguenay-Lac-St-Jean, (03) Quebec, (04) Mauricie, (05) Estrie, (06) Montreal, (07) Outaouais, (08) Abitibi-Témiscamingue, (09) Côte-Nord, (10) Nord-du-Québec, (11) Gaspésie-Îles-de-la-Madeleine, (12) Chaudière-Appalaches, (13) Laval, (14) Lanaudière, (15) Laurentides, (16) Montérégie, (17) Centre-du-Québec. The Greater Montreal area (regions 06, 13 and 16) is enlarged in the insets. Other BRCA mutations identified by yellow sections in the pies indicate other mutations identified in (01) BRCA1: 2244insA; (02) BRCA1: 2244insA and 5221delTG; (03) BRCA1: 2080insA and 2953 delGTAinsC, BRCA2: 3034delAAAC and 8904delA; (04) BRCA2: 2816insA and R3128X; (05) BRCA1: 3705insA; (12) BRCA1: 2244insA, BRCA2: 6503delTT; (15) BRCA1: 4160delAG; (16) BRCA1: 2953 delGTAinsC; (17) BRCA1: E352X; New Brunswick BRCA1: 1623delTTAAA. The Statistics Canada population census of these regions of Quebec and New Brunswick for 2001 was (01) 200 630; (02) 278 279; (03) 638 917; (04) 255 268; (05) 285 613; (06) 1 812 723; (07) 315 546; (08) 146 097; (09) 97 766; (10) 38 575; (11) 96 924; (12) 383 376; (13) 343 005; (14) 388 495; (15) 461 366; (16) 1 276 397; (17) 218 502; New Brunswick, 749 900.
Analysis of age at diagnosis for breast or ovarian cancer
In the 191 families from subset A, the mean age at diagnosis for female breast cancer was 45.1, 49.3 and 53.1 years for BRCA1-positive, BRCA2-positive and BRCA-negative families, respectively (p<0.0001; table 3). Individuals with BRCA1 mutations had a significantly earlier age at diagnosis than those with BRCA2 (p = 0.015). These results demonstrate a younger age at diagnosis for BRCA-positive families and for high-risk families (no mutation detected) compared with all the cases diagnosed in Quebec between 1993 and 1997,54 in which 54% of patients were diagnosed at age ⩾60 years, with an estimated average age at diagnosis of 61.3 years.
Comparison of age at diagnosis in 191 families with at least one affected individual tested
The average age at onset for ovarian cancer was 52.8, 56.1 and 58.5 years for BRCA1-positive, BRCA2-positive and BRCA-negative families, respectively (p = 0.04), with no significant difference between the age at diagnosis of ovarian cancer between BRCA1-positive and BRCA2-positive cases (table 3). In contrast with breast cancer, the age at diagnosis of ovarian cancer in our cohort was quite similar to that in the Quebec population.54
Prediction of BRCA1/BRCA2 carrier status based on family history
Figure 4 shows the distribution of all 256 families (subsets A and B) according to the number of breast cancer cases aged <50 years old, ⩾50 years old, the number of patients with ovarian cancer and the mutation status of the family. The number of early-onset breast cancers, the number of ovarian cancers and the presence of breast cancer in men, especially for BRCA2, seem to be predictors of the mutation status. For example, in families from subset A, the presence of ⩾2 ovarian cancers was associated with the highest risk of identifying a mutation in these families, with 18/26 (69%) of them having either a BRCA1 (n = 13) or BRCA2 (n = 5) mutation. Whereas 42% of those having one case of ovarian cancer harboured a BRCA1/BRCA2 mutation, the proportion of BRCA1-positive families was 60% compared with 40% of BRCA2-positive families.
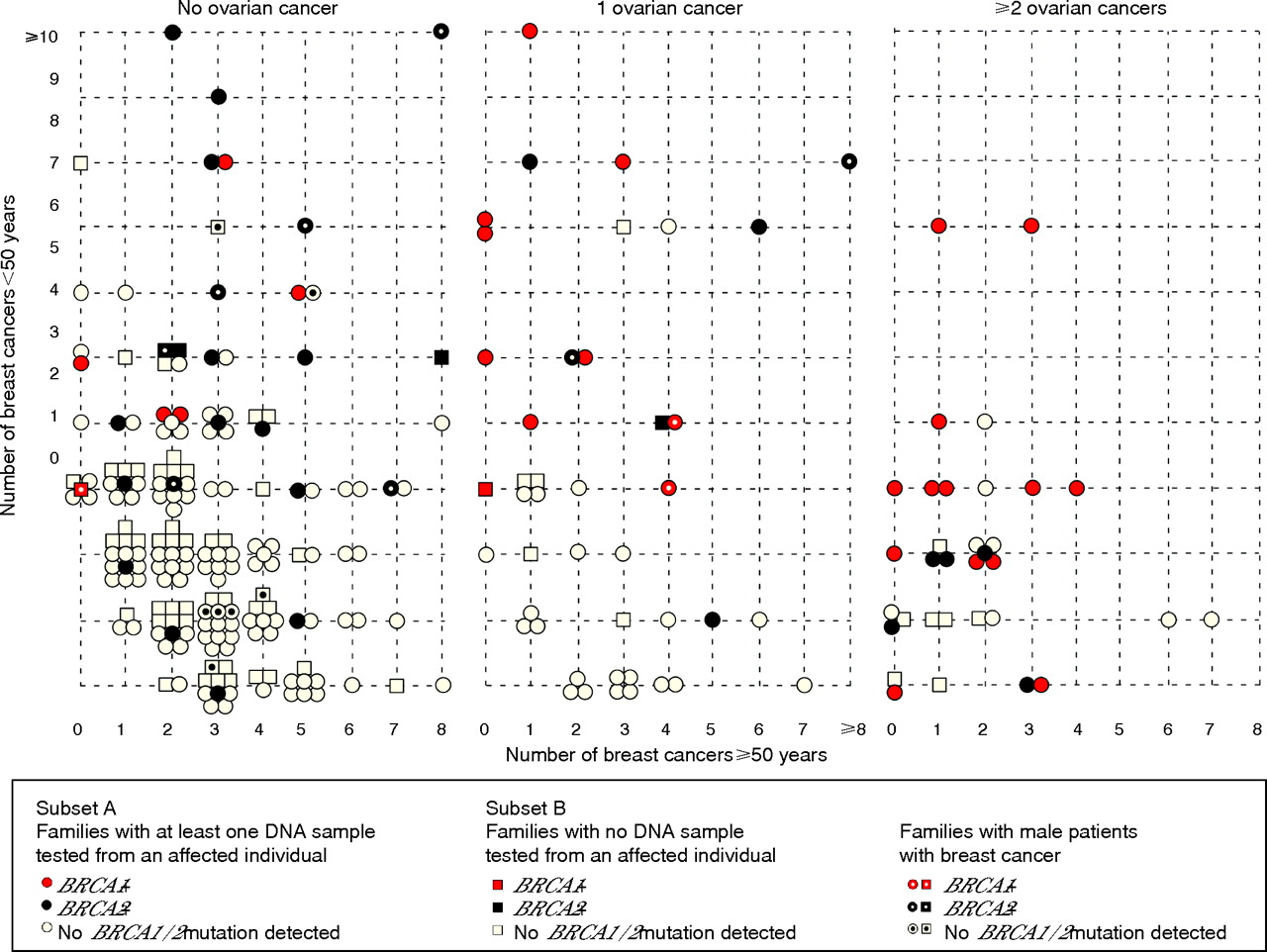
Distribution of the 256 families according to the presence of ovarian cancer, the number of breast cancer cases diagnosed before (<) or after (⩾) 50 years and their mutation status. BRCA1-positive (+) families are represented by either a red circle (families of subset A) or a red square (families of subset B). BRCA2-positive families are represented by either a black circle (families of subset A) or a black square (families of subset B). Families with no BRCA1/BRCA2 mutation detected are represented by either an open circle (families of subset A) or an open square (families of subset B). Female breast and ovarian cancer cases were at most third-degree relatives from the proband, whereas the presence of a male patient with breast cancer was considered at any degree from the proband. A dot in each form indicates the presence of male patients with breast cancer in the family.
Logistic regression was performed in the 191 high-risk families from subset A (table 4) to distinguish more precisely family history, features between BRCA1/BRCA2-positive families and families in which no BRCA1/BRCA2 mutation was identified, and to estimate the likelihood of identifying a BRCA1 or BRCA2 mutation in a French-Canadian family taking into consideration the index case’s family history of cancer (ie, all cases at most third-degree relatives from index case). This empirical predictive model indicates that the presence of at least 2 cases of ovarian cancer and ⩾4 cases of breast cancer diagnosed before 50 years are significant predictors for the presence of a segregating BRCA1/BRCA2 mutation. Table 4 also shows that the presence of breast cancer in men and bilateral breast cancer are also significant predictors of the familial BRCA1/BRCA2 mutation status.
Logistic regression for prediction of carrier status based on family history in 191 families with at least one affected individual tested
Comparison of empirical models for predicting BRCA1/BRCA2-positive families
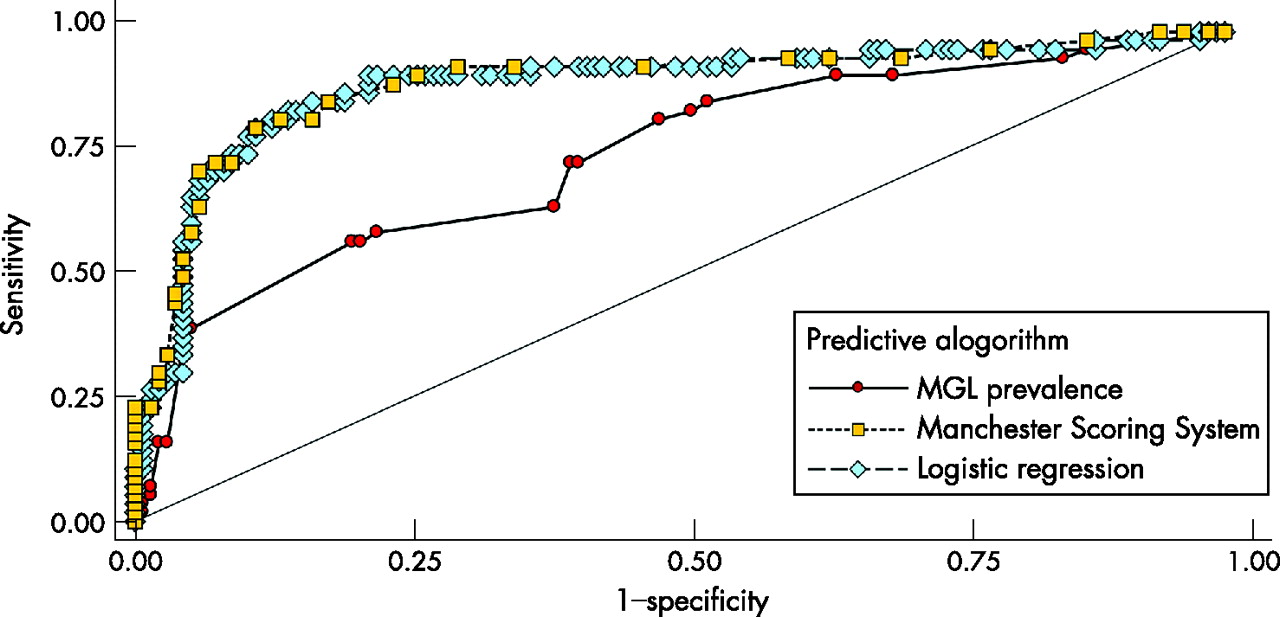
Using ROC curves, we evaluated the accuracy of our logistic regression model compared with two other empirical methods to discriminate between those families segregating a BRCA1/BRCA2 mutation and those who do not. Figure 5 shows the ROC curves for the Manchester and MGL Scores obtained using subset A data. The classifications based on the Manchester Score and the logistic regression approach are quite similar, with the tables based on mutation prevalence from the MGL dataset clearly not performing as well. The areas under the ROC curves (a common measure of the adequacy of a quantitative predictive algorithm) are 0.89 (95% confidence interval (CI) 0.84 to 0.95), 0.89 (95% CI 0.83 to 0.95) and 0.75 (95% CI 0.66 to 0.83) for the Manchester, logistic regression and MGL prevalence table approaches, respectively; the MGL prevalence table classifier is significantly lower than the other two (p = 0.0001). Finally, the ability of these models to predict accurate probabilities was examined using Brier Scores. The Brier Score for the logistic regression approach was 0.11 (p = 0.33), whereas that for the MGL prevalence was higher (Brier Score = 0.18; p<0.0001), indicating a poor fit to the observed data.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Receiver operating characteristic analysis of three BRCA1/BRCA2 mutation prediction algorithms showing sensitivity (ie, proportion of families with a BRCA1/BRCA2 mutation with a score or probability greater than or equal to a given threshold) plotted against 1−specificity (ie, proportion of families with scores below the threshold without an identified BRCA1/BRCA2 mutation). One point on the curve is obtained for each possible threshold. The area under the curve is used as a measure of the adequacy of the criteria used for genetic testing. MGL, Myriad Genetics Laboratories.
DISCUSSION
This translational research study provides important data for the evaluation of the analytical and clinical validity of predictive genetic testing for deleterious mutations in BRCA1 and BRCA2 susceptibility genes in the French-Canadian population. This study, begun 10 years ago, was designed to generate needed data about the BRCA1/BRCA2 mutation spectrum, prevalence and geographical distribution of these highly penetrant alleles in the Quebec population having a French ancestry. This key information was then coupled with the family history of cancer and selection criteria to determine the optimal strategy for clinical genetic testing in this founder population, keeping in mind the limited resources allocated to such a predictive test in a nationalised healthcare system and the necessity to target interventions at high-risk individuals who have the most health benefits to gain from available preventive and risk-reduction strategies.
BRCA1/BRCA2 mutation spectrum, prevalence and regional distribution
Given the comprehensive genetic analyses performed and the fact that almost all our participating families have a minimum of eight generations of mostly French-Canadian ancestry, this study provides for an accurate estimation of the proportion of BRCA1/BRCA2-positive and BRCA1/BRCA2-negative families with multiple cases of breast or ovarian cancer in the French-Canadian population in Quebec. Of 15 mutations identified in subsets A and B of this study, 8 mutations have never been previously reported in the French-Canadian population (E352X, 1623delTTAAA, 2080insA, 2244insA, 3705insA and 4160delAG in BRCA1; 8904delA and R3128X in BRCA2). Moreover, another study recently reported eight additional mutations that were not observed in our cohort (Q81X, W321X and 3875delGTCT for BRCA1; 2558insA, 3398delAAAAG, 3773delTT, E1953X and 7235G→A in BRCA2).25 However, when combining results of both studies, a total of 11 deleterious mutations were found to be recurrent (ie, observed in ⩾2 families) in the French-Canadian population of Quebec (2244insA, 2953delGTAinsC, 3875delGTCT, R1443X and 5221delTG in BRCA1; 2816insA, 3034delAAAC, 3398delAAAG, E1953X, 6503delTT and 8765delAG in BRCA2), 8 of which were detected in our cohort. It is also of interest to note that the frequency of the founder R1443X mutation, introduced in the 17th century,43 in our cohort (29% of all mutation-positive families and 30.3% of those positive in subset A) is similar to that found in the Oros cohort (33.8% of mutation-positive families).25
The number of families harbouring a deleterious BRCA1/BRCA2 mutation was 56 (29.3%) in subset A, in which at least one affected individual (always including the youngest affected participant) underwent full BRCA1 and BRCA2 gene sequencing after the initial targeted sequencing of a panel of 29 published or observed mutations in the French-Canadian population. The prevalence of positive families is in the range of that observed in multiple-case families in other populations,55–58 but this range is quite large and could be attributable to family inclusion criteria and to mutation detection techniques used.
To investigate whether our mutation spectrum of BRCA1 and BRCA2 in French-Canadian families with high risk of breast and/or ovarian cancer has been biased by PCR-based direct sequencing methods, the remaining 135 inconclusive families from subset A were also screened for large deletions or rearrangement using Southern blot and multiple ligation-dependent amplification analysis.40 No large deletion or duplication or other rearrangements were found in BRCA1 and BRCA2, thus suggesting that the frequency of such mutations is quite low in our population.
In addition to clearly deleterious protein-truncating mutations, a number of unclassified sequence variants were included in the panel of tested mutations or detected by sequencing during the course of this study. We believe that the vast majority of these sequence variants have no clinical relevance, but it is possible that a small number of these changes could eventually prove to be deleterious. However, this should not change the basic conclusions and testing strategy from this paper. Further studies using recently developed methods49–53 are needed to better understand the clinical relevance of these sequence variants observed in our cohort.
However, the prevalence observed in this cohort study cannot be directly compared with that observed in a previous study on French-Canadian populations because of marked differences in study design and ascertainment criteria.25 In the previous study, although the testing approaches used were less comprehensive, thus leading to an expected lower sensitivity, 44% of the families were positive. This can be explained, at least partly, by the higher propensity to ascertain families with ovarian cancer in the Oros study than in our own (44% v 32%).
Demographic reasons could also explain the differences observed in the spectrum of mutations identified in our study compared with the Oros study. Firstly, the French-Canadian origin of our participating families was confirmed by genealogical reconstruction, yielding an average depth of 10 generations of mostly French-Canadian ancestry. In the cohort described by Oros et al,25 familial origin was based on the index case reporting French-Canadian ancestry of grandparents. Moreover, their families were ascertained from medical centres in Montreal, which is located in the western part of the province. In our study, families were recruited from all regions of Quebec, but most of them (75%) were referred by clinicians working in the eastern part of the province, including the Quebec City area. Genealogical and genetic studies have shown important variability among regions in Quebec, with many studies presenting the eastern regions as those where the consequences of the initial French founder effect are most conspicuous.59–61 This phenomenon could explain the much higher frequency of the founder 8765delAG mutation found in our cohort study (45% of all mutation-positive families) than in the Oros cohort (23%). After the British conquest of 1760, most immigrants, who were of various origins, settled in the western part of the province. Moreover, there was important mobility, linked to the urbanisation process, taking place from the eastern to western regions59; these factors contributed in shaping the present-day genetic pool of the Quebec population, which is expected to be more diverse in the western part of the province. Therefore, we can hypothesise that the mutation spectrum identified in our cohort is more representative of the gene pool introduced by the founders who settled during the French regime, whereas the mutations specific to the Oros cohort would probably have more recent and more heterogeneous origins. Moreover, in the 19th and 20th centuries, the population in that territory expanded, leading to regional and local founder effects, and this expansion could have an effect on the current distribution of some mutations.
Predicting BRCA1/BRCA2-positive family status to devise an optimal multistep genetic testing approach
As BRCA1/BRCA2 mutation screening testing is expensive, estimation of familial prior probability using a model appropriate for our population is an important issue. Several tools have been developed to help the clinician in predicting the probability of carrying a BRCA1/BRCA2 mutation based on the familial history of breast and/or ovarian cancer. The Manchester Scoring System is one of the most recent tools to determine whether the likelihood of identifying a mutation in a family reaches the 10% threshold for either BRCA1 or BRCA2.33 This model is easy to use and does not require computer implementation. This scoring system was devised using a combination of results from screening and the family history of mutation-negative and mutation-positive kindreds in the Manchester region of northwest England. Our results show that the empirical Manchester Scoring System is effective in predicting the likelihood of the presence of a mutation in French-Canadian families. For instance, our analysis in subset A showed that the probability of identifying a mutation in either the BRCA1 or BRCA2 gene, if complete sequencing is performed in at least one affected individual from such multiple-case families, was 4% (4/99), 20% (2/10), 23% (5/22), 56% (9/16) and 82% (36/44) at ranges of scores 4–14, 15–16, 17–20, 21–24 and ⩾25, respectively. Further, our ROC curve analysis also indicated that a cut-off at a Manchester Score of 18 seems optimal as a threshold (ie, a sensitivity of 86%, a specificity of 82%, a positive predictive value of 66% and a negative predictive value of 93%) for predicting the BRCA1/BRCA2-positive family status. In other words, among the 73 families showing scores ⩾18, 48 were BRCA1/BRCA2 positive, but by testing only these families, 8 families carrying a recurrent BRCA1/BRCA2 mutation would be missed. Moreover, our ROC curve analysis also showed that using a cut-off of 15 will predict correctly the presence of a mutation in 52 families among the 95 families reaching this threshold (ie, a sensitivity of 93%, a specificity of 68%, a positive predictive value of 55% and a negative predictive value of 96%). Our finding is thus in accordance with the update recently published by Evans et al34 and with the results observed in a recent Danish study.62
Surprisingly, the Manchester Scoring System performed equivalently to the approach based on logistic regression models developed on the same families used for the prediction. The predictive power and general characteristics of the Manchester model based on the UK population fit well with those found in French-Canadian families despite the quite different ascertainment criteria and the founder effect in our population, which shows quite well the robustness of this relatively simple approach. This is in contrast with the poorer performance of the MGL prevalence tables, which may indicate the effect of the lack of strict personal and family history requirements for testing in the US. We have previously shown that the genetic model Breast and Ovarian Analysis of Disease Incidence and Carrier Estimation Algorithm, developed using data from the UK, accurately predicts the number of BRCA1 and BRCA2 mutations for various groups of families and discriminates well at the individual level between carriers and non-carriers.29
The presence of a founder effect in the French-Canadian population and the higher prevalence of few recurrent BRCA1 and BRCA2 mutations provide the opportunity to evaluate a panel of mutations that could be used as an initial screening test in the framework of a multistep testing approach. The sensitivity (here defined to mean the proportion of all mutation-positive individuals detected) of a genetic test based on the two most common mutations, R1443X and 8765delAG, was 75%, with a negative predictive value at 91%, whereas the sensitivity of a genetic test based on the eight recurrent mutations was 88%, with a negative predictive value of 95% in this study. When combining our data with those from the literature, testing for the presence of the 11 recurrent mutations (2244insA, 2953delGTAinsC, 3875delGTCT, R1443X and 5221delTG in BRCA1; 2816insA, 3034delAAAC, 3398delAAAG, E1953X, 6503delTT and 8765delAG in BRCA2) should be considered to be a cost-effective initial test of individuals from high-risk French-Canadian families in Quebec. Considering the relatively low cost of such an initial test, the utility of inclusion in this panel of any additional recurrent mutation that will be observed in the future in ⩾2 French-Canadian families should be examined.
Prediction models described above can help to categorise families in which no mutation was found by such an initial test, but possess a high prior probability to harbour a BRCA1/BRCA2 mutation detectable by a more comprehensive testing approach. In this regard, to further examine the usefulness of the Manchester Scoring System for this purpose, we performed ROC curve analysis using data from only the 143 families from subset A in which no BRCA1/BRCA2 mutation was detected after testing the panel of recurrent French-Canadian mutations. This analysis indicates that using Manchester Scores ⩾18 as a threshold predicts the presence of a BRCA1/BRCA2 mutation in this subset of families, with a sensitivity of 100%, a specificity of 82% and a positive predictive value of 25% (data not shown). In other words, among the 32 families showing a score of ⩾18, the second-step testing approach (full sequence analysis) led to the discovery of all 8 families harbouring a deleterious mutation, thus giving the possibility to avoid performing this second-step test in 111/143 (78%) families. Alternatively, by selecting all families with a score of ⩾15 (n = 48), the specificity of the second-step test will be 70.4% and the positive predictive value will be 16.7% without decreasing its sensitivity. This threshold can be adapted in relation to clinical setting characteristics and judgement of health professionals about the appropriateness of further testing based on both the personal and familial history of cancer of their patients.
Although it is generally preferable to test affected individuals, this is not always possible in clinical practice, especially given the poor prognosis of ovarian cancer and of some early-onset high-grade breast cancers affecting BRCA1/BRCA2 carriers. Moreover, such a requirement may also raise some ethical and psychosocial issues. On the other hand, unaffected individuals at high genetic risk, generally young women, are those who would benefit the most from such genetic testing. Our analysis of subset B of high-risk families in which only asymptomatic individuals were tested for the panel of French-Canadian mutations indicates that almost 10% of those high-risk families harboured BRCA1/BRCA2 mutations. All six BRCA1/BRCA2-positive families had Manchester Scores ⩾16 (16, 21, 24, 27, 27 and 35), which were indicative of a higher prior probability to segregate such a mutation. Only 3 of these BRCA1/BRCA2-positive families were found among the 45 families in which only one unaffected first-degree relative of an affected family member was tested, whereas the 3 other positive families were found among the 15 families in which ⩾2 unaffected first-degree relatives were tested, including one man with prostate cancer diagnosed at 52 years of age. Indeed, the overall likelihood of identifying a mutation under such a test in asymptomatic individuals is dependent on several parameters, including family history, type of cancers, age at onset, ethnic background and, especially, the number and degree of kinship of unaffected relatives of an affected family member being tested as well as the sensitivity of the test offered.63 Several risk-prediction methods can be used to estimate the carrier probability of asymptomatic individuals, in order to design an optimal testing strategy for those families without an affected relative available.30
Considering the ascertainment criteria of our cohort study targeting high-risk families, it was not surprising to observe that families having Manchester Scores ⩾4 had a 25.7% probability of harbouring a deleterious BRCA1/BRCA2 mutation detectable by this initial test, but a 29.3% probability of carrying a mutation detectable by a comprehensive test, thus supporting that it was appropriate to recommend such families for testing, in agreement with several international guidelines64. Recommended probability thresholds for BRCA genetic testing vary widely according to available resources or other considerations, ranging from 5–7% recommended by the US Department of Defense to ⩾20% by the UK National Institute for Health and Clinical Excellence guidelines. In Canada, a cut-off of >10% probability has been proposed in the Ontario testing guidelines.65
Taking into consideration the recommendations described above and our findings, it seems reasonable in a clinical setting to offer to individuals with a personal or family history of cancer, with a lower restrictiveness, to be initially tested for the panel of recurrent mutations in the French-Canadian population. Prediction models described above can help clinicians in evaluating the appropriateness to offer further testing for those families in which no mutation was found at the initial test, but possess a high prior probability of harbouring a BRCA1/BRCA2 mutation detectable by a more comprehensive testing approach, such as those having Manchester Scores ⩾18. Lower thresholds could be used as resources become available in the healthcare system and where the judgement of healthcare professionals involved in familial cancer clinics so indicates.
Acknowledgments
We thank the participants and their families for participating in this study. We thank the clinicians from the province of Quebec for participant referral, especially Drs Martine Aubry, Marie-Claude Audet, Christiane Bouchard, Luc Deschênes, Gaston Dorval, Pierre Dupont, Michel Fortier, Sylvain Gagnon, Francine Lévesque, Louise Morin, Hélène Otis and Hélène Savard who referred more than five high-risk families to this translational research study. We also thank Dr Sylvie Délos, Nathalie Bolduc, Pascale Léger, Andrée McMillan, Geneviève Ouellette and Tina Babineau of the Cancer Genomics Laboratory (Quebec) for sample management and skilful technical assistance, Michèle Jomphe from the BALSAC Project (Chicoutimi) as well as Lise Gobeil from the Interdisciplinary Research Group on Demography and Genetic Epidemiology (Chicoutimi) for genealogical reconstruction and analyses, and Laurent Richard and Nicolas Lanouette from the Historical Geography Laboratory at Laval University for cartography work. We also greatly appreciate the advice received from the different ethics committees.
REFERENCES
Footnotes
-
↵* These authors contributed equally to this work and should be considered as the first author.
-
Published Online First 11 August 2006
-
Competing interests: The authors declare no competing interests.
-
Funding: This work was supported by the Canadian Institutes of Health Research (CIHR) and their Institute of Cancer and Institute of Gender and Health for the INHERIT BRCAs research programme, Fonds de la Recherche en Santé du Québec (FRSQ)/Réseau de Médecine Génétique Appliquée (RMGA), the Canadian Breast Cancer Research Alliance and the CURE foundation. JS is chairholder of the Canada Research Chair in Oncogenetics.
-
Other members of INHERIT BRCAs involved in this study are as follows: Marc Tremblay, Interdisciplinary Research Group on Demography and Genetic Epidemiology (GRIG), University of Quebec at Chicoutimi, Chicoutimi, Canada; Olga Sinilnikova, International Agency for Research on Cancer (IARC), Lyon, France, and Plate-forme de Génétique Constitutionnelle des Cancers Fréquents, Hospices Civils de Lyon/Centre, Léon Bédard, Lyon, France; Antonis Antoniou, CRC Genetic Epidemiology Unit Strangeways Research Laboratories, University of Cambridge, UK; and Michel Dugas, Département de psychologie, Concordia University & Centre de recherche de l’Hôpital du Sacré-Cœur, Montreal, Canada.
Linked Articles
- Miscellaneous