Article Text

Abstract
Background DNA replisome is a molecular complex that plays indispensable roles in normal DNA replication. IMAGE-I syndrome is a DNA replisome-associated genetic disease caused by biallelic mutations in the gene encoding DNA polymerase epsilon catalytic subunit 1 (POLE). However, the underlying molecular mechanisms remain largely unresolved.
Methods The clinical manifestations in two patients with IMAGE-I syndrome were characterised. Whole-exome sequencing was performed and altered mRNA splicing and protein levels of POLE were determined. Subcellular localisation, cell cycle analysis and DNA replication stress were assessed using fibroblasts and peripheral blood from the patients and transfected cell lines to determine the functional significance of POLE mutations.
Results Both patients presented with growth retardation, adrenal insufficiency, immunodeficiency and complicated diffuse large B-cell lymphoma. We identified three novel POLE mutations: namely, a deep intronic mutation, c.1226+234G>A, common in both patients, and missense (c.2593T>G) and in-frame deletion (c.711_713del) mutations in each patient. The unique deep intronic mutation produced aberrantly spliced mRNAs. All mutants showed significantly reduced, but not null, protein levels. Notably, the mutants showed severely diminished nuclear localisation, which was rescued by proteasome inhibitor treatment. Functional analysis revealed impairment of cell cycle progression and increase in the expression of phospho-H2A histone family member X in both patients.
Conclusion These findings provide new insights regarding the mechanism via which POLE mutants are highly susceptible to proteasome-dependent degradation in the nucleus, resulting in impaired DNA replication and cell cycle progression, a characteristic of DNA replisome-associated diseases.
- DNA replication
- immune system diseases
- mutation
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
IMAGE-I syndrome is caused by compound heterozygous mutations of intronic and exonic loss-of-function mutations in DNA polymerase epsilon catalytic subunit 1 (POLE) gene. However, whether other mutations can cause IMAGE-I syndrome and underlying molecular mechanisms remain unclear.
WHAT THIS STUDY ADDS
We identified a unique deep intronic mutation and novel two exonic loss-of-function mutations in POLE gene in two Japanese patients with IMAGE-I syndrome. The mutations resulted in impaired DNA replication and cell cycle progression caused by reduced, but not null, protein expression. The POLE mutants showed severely diminished nuclear localisation by proteasome-dependent degradation.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE AND/OR POLICY
This study reinforces the molecular mechanisms that a characteristic combination of a deep intronic mutation and loss-of-function mutation causes IMAGE-I syndrome in humans. Aberrant subcellular localisation of POLE mutants provides us with new insights regarding the quality control of POLE functions in the nucleus.
Introduction
DNA replication is a fundamental process required for cell proliferation and differentiation. Multiple factors tightly regulate this process, and over the past decade, their impairment has been known to affect growth and development in humans.1 Germline mutations associated with DNA replication have been identified in patients with intrauterine growth retardation (IUGR), and short stature are often complicated by adrenal insufficiency and immunodeficiency. IMAGe syndrome (MIM number: 600856), which stands for IUGR, metaphyseal dysplasia, adrenal hypoplasia congenita and genital anomalies, is the most well-known disease in this category and is caused by gain-of-function variants in the maternal CDK1NC allele.2 Other abnormalities in factors related to cell proliferation, such as GINS1 deficiency (610608),3 MCM4 deficiency (609981)4 and MIRAGE syndrome caused by functional gain mutations in SAMD9 (610456) result in similar phenotypes.5
DNA polymerase epsilon is an essential polymerase involved in the synthesis of DNA leading strands in eukaryotes and comprises four subunits. Mutations in POLE (GenBank: NM_006231.3), which encodes DNA polymerase epsilon catalytic subunit 1, the largest subunit of the polymerase, was reported by Logan et al 6 to cause IMAGe syndrome with immunodeficiency (IMAGE-I syndrome, MIM number: 618336).6 POLE has been associated with FILS syndrome (615139), which is characterised by facial dysmorphism, immunodeficiency, livedo and short stature.7 8 These disorders are associated with unique mutations, such as the biallelic mutation of c.4444+3A>G in FILS syndrome, and compound heterozygous mutations of intronic c.1686+32C>G and exonic loss-of-function mutations in IMAGE-I syndrome. The intronic mutations are possibly important for POLE-associated diseases, as these variants cause partial splicing alterations. However, more reports regarding this are lacking, and whether other mutations can cause IMAGE-I syndrome remains unclear.
Here, we report the cases of two Japanese patients with IMAGE-I syndrome. We identified novel intronic and exonic POLE mutations and investigated whether these mutations play a significant role in the molecular mechanisms underlying the disease.
Methods
Natural killer (NK) cell cytotoxic activity
NK cell cytotoxic activity was measured by 51Cr-release assay performed by SRL in Japan.9 The reference standard value is 18%–40%.
Genetic analysis
Genomic DNA was extracted from whole blood cells using the PAXgene blood DNA kit (Qiagen, 761133). Total RNA was extracted from whole blood cells or fibroblasts using the RNeasy Plus mini kit (Qiagen, 74134), and cDNA was generated using the PrimeScript RT reagent kit with gDNA eraser (TaKaRa, RR047A). Whole-exome sequencing and Sanger sequencing were performed as described previously.10 The primers used for RT-PCR and Sanger sequencing are listed in online supplemental table 1.
Supplemental material
POLE expression vectors
Full-length wild type (WT) and mutant POLE cDNA harbouring c.711_713del and four synonymous single nucleotide variants were amplified from fibroblasts of healthy control and patient 1, respectively, and cloned into pCMV6-plasmid with a FLAG tag at the C-terminus (Origene, PS100001). The Myc-tag was removed when the linearised pCMV6 vector was amplified via PCR using the In-fusion HD cloning kit (TaKaRa, 639648). Other POLE mutants (c.2593T>G and c.3019G>C) were generated via site-directed mutagenesis. All constructs were verified using Sanger sequence.
Minigene assay
POLE minigene fragments, from exon 10 to intron 13, were amplified from the genomic DNA of healthy control and cloned into the pCMV4 plasmid (Addgene, 21966). The c.1226+234G>A mutation was introduced via site-directed mutagenesis. HEK-293T cells were transfected with the plasmids and pCMV4 empty vector using Lipofectamine LTX plus reagent (Thermo Fisher Scientific, 15338100) and incubated for 24 hours. Total RNA was extracted, followed by RT-PCR and 1.5% agarose gel electrophoresis.
Immunoblotting
Total protein was extracted from fibroblasts cultured at 80% confluence or peripheral blood mononuclear cells (PBMCs). PBMCs were isolated from whole blood using Ficoll Paque Plus (Cytiva, 17144002) and stimulated with haemagglutinin (10 µg/mL) and interleukin 2 (0.1 µg/mL) to induce POLE expression. These cells were lysed with the LysoPure nuclear and cytoplasmic extractor kit (Wako, 295–73901). Protein concentration was estimated using the Pierce bicinchoninic acid protein assay kit (Thermo Fisher Scientific, 23227), and equal amounts of protein were resolved using sodium dodecyl sulfate-polyacrylamide gel electrophoresis in a Mini-PROTEAN TGX 4%–15% stain-free gel (Bio-Rad, 4568085) and transferred to a polyvinylidene difluoride membrane. The membrane was probed with antibodies against POLE (GeneTex, GTX132100), FLAG (Sigma-Aldrich, F1804), lamin A/C (Santa Cruz, 123243) and α-tubulin (Santa Cruz, 24534) followed by incubation with appropriate horseradish peroxidase-conjugated secondary antibodies. The bands were detected using the ECL Prime western blotting system (Cytiva, GERPN2232) and ChemiDoc MP system (Bio-Rad), and quantified using the Image Lab software normalising to total protein content using the stain-free gel system (Bio-Rad).
Immunofluorescence staining
COS-7 cells (1.0×104/well) were transfected with pCMV6-POLE plasmids (WT, c.711_713del, c.2593T>G and c.3019G>C) and cultured for 48 hours in an eight-well chamber slide (Matsunami, SCC-008). The cells were treated with cycloheximide (CHX) and MG132 for 5 hours before harvesting. Then, the cells were fixed with 4.0% paraformaldehyde for 10 min and permeabilised with 0.3% Triton X-100 in phosphate-buffered saline (PBS) for 10 min. To suppress the non-specific binding of antibodies, Blocking One Histo (Nacalai Tesque, 06 349–64) was added and incubated for 1 hour. The antibodies were diluted with 5% Blocking One Histo in PBS-Tween 20, and the cells were probed with anti-Flag M2 antibody (Sigma-Aldrich, F1804) or anti-phospho-H2A histone family member X (H2AX) (Ser139) antibody (Cell Signaling, 2577) for 1 hour, followed by incubation with donkey anti-mouse or anti-rabbit IgG conjugated with Alexa Fluor 568 (Abcam, ab175700 or ab175692). Subsequently, Alexa Fluor 488 phalloidin (Thermo Fisher Scientific, A12379) was added and incubated for 1 hour to stain polymerised actin. The nuclei were stained with ProLong Gold antifade mountant with DAPI (Thermo Fisher Scientific, P36935). Fluorescent images were obtained using BZ-X810 (Keyence). The signals were quantified using the ImageJ software. In total, 20 pictures at 20× magnification were captured for analysis, and 200–300 cells for each sample were examined.
Cell cycle analysis
Fibroblasts from patients and normal controls were seeded in 10-cm dishes and pulse labelled with 5-ethynyl-2-deoxyuridine (EdU) (final concentration, 10 µM) and incubated for 1 hour. After washing with PBS, the cells were incubated for 0, 4, 8 and 12 hours in fresh medium, and harvested using trypsin/EDTA and centrifuged. The cell pellets were permeabilised with 0.1% TritonX-100 in 0.1% sodium citrate for 7 min and washed with 1% bovine serum albumin (BSA) in PBS and centrifuged. This was followed by the addition of the Click-iT reaction cocktail of the Click-iT plus EdU Alexa Fluor 647 flow cytometry assay kit (Thermo Fisher Scientific, C10634). The samples were incubated for 30 min. After washing with 1% BSA in PBS and centrifugation, the cells were incubated with FxCycle propiodium iodide (PI)/RNase staining solution (Thermo Fisher Scientific, F10797) for 30 min and analysed using FACS Canto Ⅱ (BD Bioscience). The gating strategy is illustrated in online supplemental figure 1.
Statistical analysis
Statistical analyses were performed with EZR V.1.51.11 P value <0.05 was considered statistically significant.
Results
Clinical characteristics of patients
Patient 1 (P1) was an 11-year-old girl who was born with IUGR in a non-consanguineous Japanese family. She was born at 41 weeks and 6 days of age with birth weight of 2352 g (−2.8 SD) and birth height of 48.0 cm (−1.3 SD) and had severe newborn jaundice. She had NK cell cytopaenia (73 /µL, 1.4% of PBMCs) and low NK cell cytotoxic activity (6 %) and was hospitalised seven times with severe viral and bacterial infections by the age of 2 years. She had skin pigmentation at birth and high adrenocorticotropic hormone (ACTH) levels. Her cortisol levels did not increase in response to the ACTH loading test, and she was evaluated to have adrenal insufficiency due to ACTH resistance, because of which cortisol replacement therapy was started. She gained motor development by holding her neck up at 5 months, sitting without support at 7 months, hands-and-knees crawling at 14 months and standing with assistance at 18 months of age. She had delayed mental development and her IQ was 33 at 9 years of age according to Tanaka-Binet intelligence scale, showing that she had severe intellectual disability. At 2 years of age, she had growing right cervical lymphadenopathy and excisional biopsy revealed Epstein-Barr virus (EBV)-positive diffuse large B-cell lymphoma (DLBCL). The DLBCL was resistant to combined chemotherapy containing rituximab. Currently, she is alive and is free from relapse following allogeneic umbilical cord blood transplantation for more than 7 years.
Patient 2 (P2) was a 31-year-old man who was born with IUGR and jaundice in a non-consanguineous Japanese family. He was born at 41 weeks and 4 days of age with a birth weight of 2162 g (−3.6 SD). He had anaemia and received red blood cell transfusion at the age of 1 month. He also presented NK cell cytopaenia (89 /µL, 3.6% of PBMCs) and low NK cell cytotoxic activity (5 %). He had growth retardation due to growth hormone insufficiency diagnosed by some loading tests and anterior pituitary atrophy in MRI and was treated with growth hormone replacement therapy. At 17 years of age, the patient developed EBV-positive DLBCL in his nasopharynx. He achieved complete response with combined chemotherapy containing rituximab and has been free from relapse for more than 13 years. He was also medicated with cortisol at the age of 22 years because of skin pigmentation, high ACTH levels and no response to the ACTH loading test.
Figure 1A shows the pedigrees of both patients and their families. P1 and P2 had similar clinical features as that observed in IUGR, facial dysmorphism (figure 1B), growth retardation (figure 1C), developmental delay, adrenal insufficiency and immunodeficiency. A summary of haematological and immunological data in P1 and P2 is shown in online supplemental table 2. Table 1 shows a summary of clinical manifestations in previously reported cases and in the two patients of this study with IMAGE-I syndrome.6
Summary of clinical manifestations and POLE mutations in previously reported cases and patients with IMAGE-I syndrome in this study

Novel POLE mutations and clinical manifestations in two patients with IMAGE-I syndrome. (A) Pedigrees of two unrelated kindreds, showing allelic segregations. ‘E?’ indicates an unknown genotype. The scheme below shows the location of the mutations in POLE. (B) Facial appearances of Patient 1 (P1) at 11 years of age and Patient 2 (P2) at 31 years of age. (C) Growth curve of P1. The upper curve shows the body height for ages, and the lower curve shows the body weight for ages. These were described using the clinical tool provided by the Japanese Society for Pediatric Endocrinology (http://jspe.umin.jp/medical/chart_dl.html). Exo, exonuclease domain; Pol, polymerase domain; POLE, gene encoding DNA polymerase epsilon catalytic subunit 1.
Identification of pathogenic variants in POLE
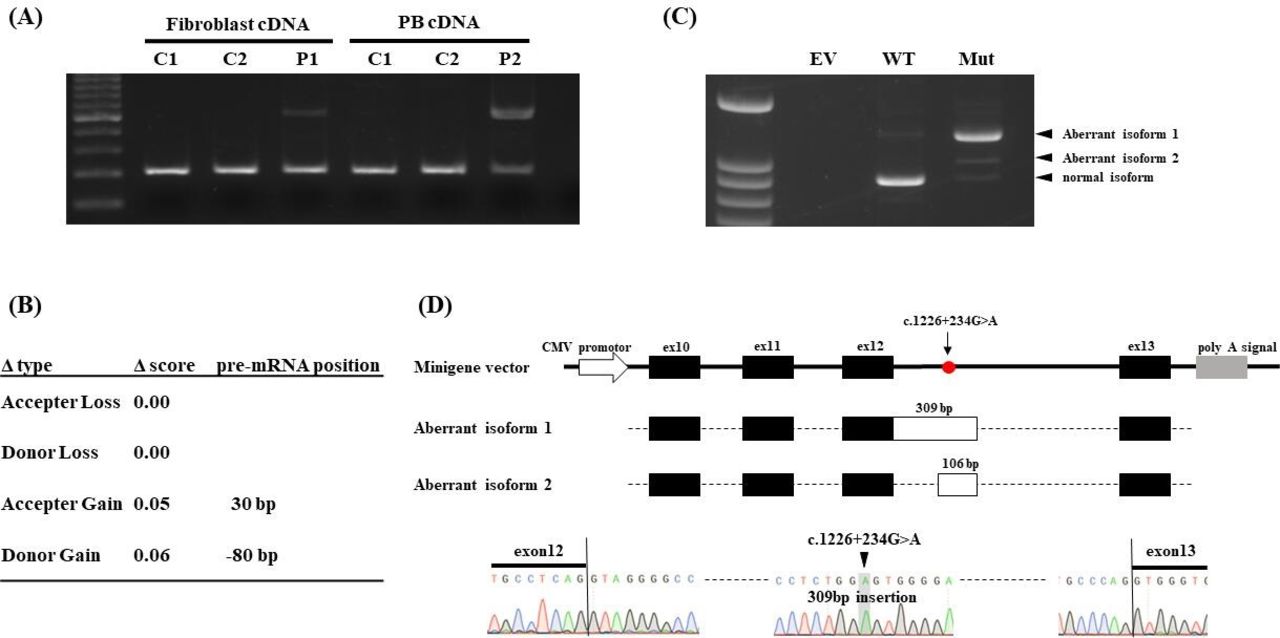
We identified a monoallelic POLE mutation in each patient using whole-exome sequencing; however, no other candidate DNA replication factors, including CDKN1C, GINS1, MCM4 and SAMD9, were mutated. P1 harboured a variant of c.711_713del (p.Lys237_Ile238delinsAsn), which has not been reported in any database. P2 harboured c.2593T>G (p.Trp865Gly, CADD score 26.9), which was not reported in gnomAD (genome aggregation database, v2.1.1), but its minor allele frequency (MAF) was reported to be 0.0001 in the Japanese population according to the ToMMo (Tohoku Medical Megabank Organization, 8.3KJPN) database. As a deep intronic splicing variant, c.1686+32C>G, has been reported to be associated with IMAGE-I syndrome, we assumed that the patients might harbour unknown splicing variants. RT-PCR analysis of all exon–exon junctions revealed that a 309-bp insertion between exons 8 and 14 was present in both patients (online supplemental figure 2). RT-PCR analysis confirmed the predominant 309-bp insertion between exons 12 and 13 in both patients (figure 2A). Re-examination of the whole-exome sequencing data identified a rare variant in intron 12, c.1226+234G>A (ToMMo MAF 0.0001, CADD 4.145). Although the SpliceAI lookup platform10 did not predict that this deep intronic mutation produced aberrant splicing isoforms due to low Δ score (figure 2B), the minigene assay indicated that the c.1226+234G>A variant produced aberrantly spliced mRNA with 309-bp insertion predominantly and 106-bp insertion in a lesser extent of POLE (figure 2C). The aberrantly spliced isoforms gained a premature stop codon and could be targeted for non-sense-mediated RNA decay (figure 2D). Thus, we identified a novel pathogenic deep-intronic mutation in POLE.

A deep intronic mutation of c.1226+234G>A causes aberrant splicing of POLE (gene encoding DNA polymerase epsilon catalytic subunit 1). (A) Electrophoresis of RT-PCR products between exons 12 and 13. Patient 1 (P1) and Patient 2 (P2) have the same aberrant splicing product. DNA ladder marker was shown in left. (B) Prediction of c.1226+234G>A mutation using the SpliceAI lookup platform. Δ score ranged from 0 to 1 and provided 0.2 (high recall), 0.5 (recommended) and 0.8 (high precision) as the cut-off values. Aberrant isoform 1 cannot be predicted using the predictive method. (C) Electrophoresis of RT-PCR products in minigene assay. The c.1226+234G>A mutant produced the two aberrant splicing products with 309-bp (aberrant isoform 1) and 106-bp insertions (aberrant isoform 2). DNA ladder marker was shown in left. (D) Scheme of minigene vector and aberrant splicing products. Both aberrant splicing products were cloned and confirmed using Sanger sequencing. PB, peripheral blood; EV, empty vector; Mut, c.1226+234G>A mutant; WT, wild type.
Low POLE levels in patient samples
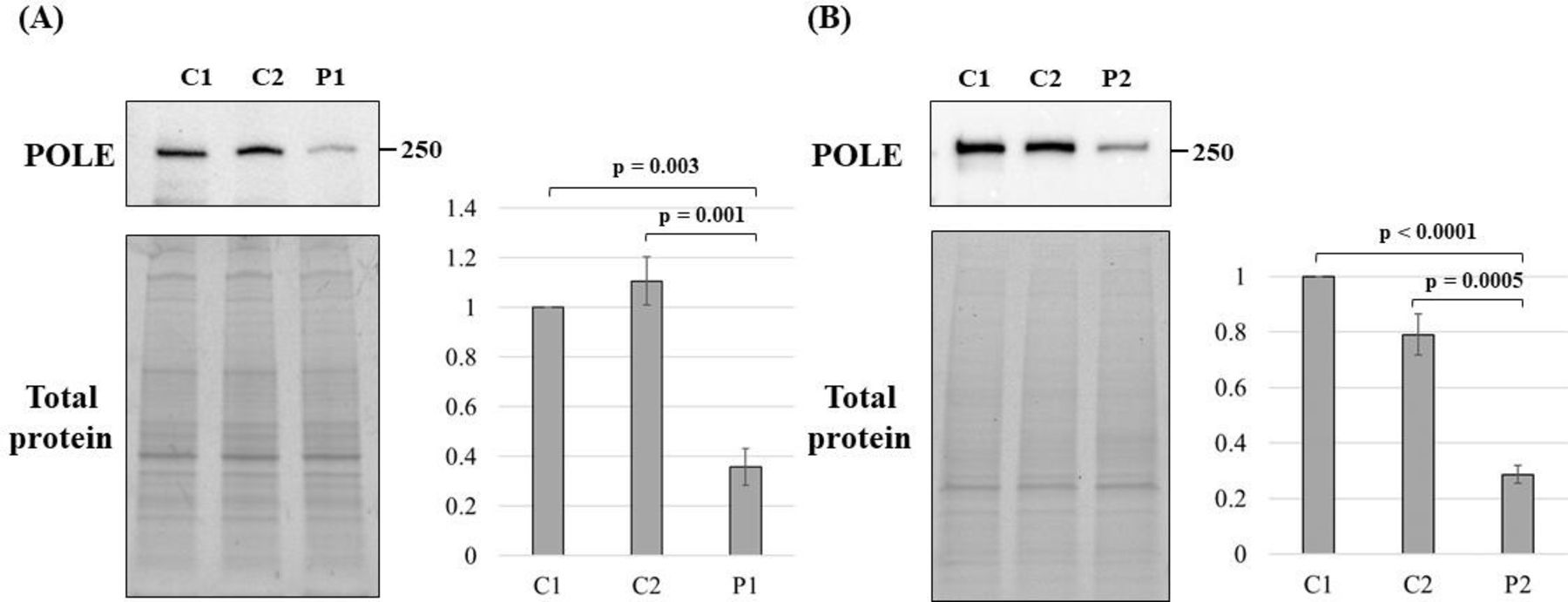
To examine protein expression levels of POLE, we performed immunoblotting of fibroblasts from P1 and PBMCs from P2. POLE levels in patient samples were significantly lower, but not null, than these from normal controls in both patients (figure 3).

POLE levels in patient samples. Immunoblotting of POLE protein extracted from two normal controls (C1 and C2) and P1 primary fibroblasts (A), and from C1, C2 and P2 PBMCs (B). The experiment was performed in triplicate and the means were calculated. The error bars indicate ±SE of the mean. The p-value was calculated using one-way ANOVA, and the Dunnett test was used for post hoc analysis. ANOVA, analysis of variance; C1, control 1; C2, control 2; P1, patient 1; P2. patient 2; PBMCs, peripheral blood mononuclear cells; POLE, gene encoding DNA polymerase epsilon catalytic subunit 1.
Impairment of cell cycle progression
As POLE insufficiency affects DNA replication, we assessed the effect of the mutants on cell cycle progression. We observed the dynamics of mid-S phase cells over 12 hours in fibroblasts from P1 (figure 4A) and PBMCs from P2 (figure 4B). Additionally, we quantified the ratio of mid-S phase cells after every 4 hours. Both line charts showed that the ratios of mid-S phase cells remained significantly higher in both patients than in normal controls, indicating that cell cycle progression was impaired in the patient cells.

Impaired cell cycle progression and replication stress response in two patients with IMAGE-I syndrome. (A), (B) FACS plot (left panels) and quantification of mid-S-phase cells (right panels). In FACS plots, each P4, P5, P6 gate shows G1/G0 phase, S phase, G2/M phase, respectively. P1 primary fibroblast (A) or P2 PBMCs (B) and three normal controls were treated with EdU for 1 hour and harvested at 0, 4, 8 and 12 hours. We performed experiments in triplicate and calculated the means. The error bars show means±SEM. The p-value was calculated using two-way ANOVA, and the Holm test was used for post hoc analysis. (C) Immunofluorescence staining of phospho-H2AX in P1 primary fibroblast and two normal controls is shown in red, and merged images with DAPI shown in blue (upper panels). Dots plots show quantification of means per cell (lower panel). The blue bars indicate the means. The p-value was calculated using the Kruskal-Wallis test, and the Mann-Whitney U test was used for post hoc test analysis. ANOVA, analysis of variance; EdU, 5-ethynyl-2-deoxyuridine; HU, hydroxyurea; P1, patient 1; P2. patient 2; PBMCs, peripheral blood mononuclear cells.
We next evaluated DNA damage using immunofluorescence for phospho-H2AX in the nucleus, as delayed DNA replication leads to increased sensitivity to DNA double-strand breaks (DSBs). H2AX is phosphorylated at DSBs, which is strongly induced when cells are exposed to hydroxyurea (HU).12 We quantified the signal per cell under stable conditions. The increase in phosho-H2AX-positive cells was statistically significant in P1 fibroblasts compared with that in normal controls (figure 4C), indicating that P1 fibroblasts were exposed to DNA replication stress, which delayed cell cycle progression.
Reduction in nuclear localisation of POLE mutants
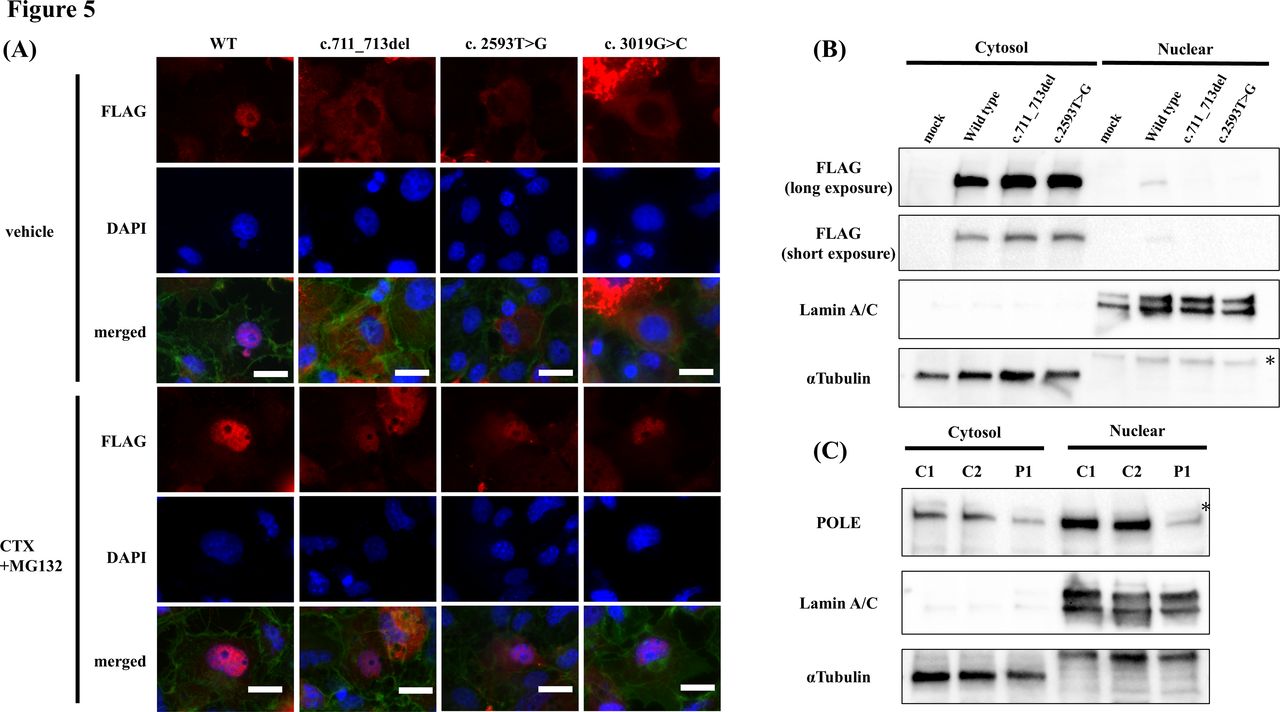
We examined the subcellular localisation of the mutants and compared it to that of the WT POLE protein to evaluate the pathogenicity of c.711_713del and c.2593T>G. Immunofluorescence staining revealed that the two overexpressed mutant POLE proteins were mainly localised in the cytosol, whereas the WT POLE was localised in the nucleus. Furthermore, we created the c.3019G>C (p.Ala1007Pro) mutant of the sole missense variant recognised as a loss-of-function mutation in a previous report (No. 10 in table 1). This mutant was also observed in the cytosol, similar to that observed for our two independent mutants (figure 5A, upper panels). The staining pattern was shown in online supplemental figure 3. Immunoblotting of the cytosolic and nuclear fractions of transfected cells also showed that POLE was present in the nuclear fraction in WT but not in the mutant of c.711_713del or c.2593T>G (figure 5B). A similar result was obtained in P1 fibroblasts, showing that the ratio of POLE expression levels in the nuclear fraction was lower than that in the normal controls (figure 5C). These results indicated that c.711_713del, c.2593T>G and c.3019G>C mutations are pathogenic and reduce nuclear localisation.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Reduced nuclear localisation of POLE mutants due to proteasome-dependent degradation. (A) Immunofluorescence staining of transfected COS-7 cells treated with vehicle (upper panels) and with CHX and MG132 (lower panels). FLAG-tagged POLE protein stained with anti-FLAG antibody is shown in red. DAPI is shown in blue, and polymerised actin stained with phalloidin is shown in green. Scale bar, 20 µm. (B) Immunoblotting of POLE in cytosolic and nuclear fraction extracted from COS-7 cells transfected with each plasmid. Introduced FLAG-tagged POLE protein was detected using anti-FLAG antibody. Lamin A/C is the marker of nuclear fraction, and α-tubulin of the cytosol fraction. *Denotes non-specific bands. (C) Immunoblotting of POLE in cytosolic and nuclear fractions extracted from P1 and two control fibroblasts. *Denotes non-specific bands. C1, control 1; C2, control 2; CHX, cycloheximide; P1, patient 1; POLE, gene encoding DNA polymerase epsilon catalytic subunit 1; WT, wild type.
Degradation of mutant POLE protein in the nucleus
Next, we next investigated the mechanisms via which nuclear localisation was reduced in these mutants. The c.711_713del mutation was located in a highly conserved region. A sequence similar to the proline-tyrosine nuclear localisation signal, a well-known nuclear localisation motif was found near the c.711_713del (online supplemental figure 4).13 The transfected cells were treated with CHX to inhibit protein synthesis and the proteasome inhibitor, MG132, to determine whether the mutant proteins were unstable and susceptible to protein degradation in the nucleus. Nuclear localisation of all three mutants was rescued by CHX and MG132 treatment (figure 5A, lower panels). Thus, these mutants were unstable in the nucleus because of proteasome-dependent degradation.
Discussion
In this study, we identified three novel POLE mutations in two patients with IMAGE-I syndrome. The deep intronic mutant generated aberrant splicing products, and pathogenic missense and in-frame deletion mutations caused protein instability in the nucleus, resulting in impaired cell cycle progression and replication stress response.
Our findings are consistent with those of a previous study in which patients with IMAGE-I syndrome harboured a deep intronic mutation. POLE is essential for cell proliferation and complete loss of its function is lethal; however, haploinsufficiency alone does not affect health because the number of loss-of-function mutations of POLE observed in the gnomAD database is similar to expected one (probability of being loss-of-function intolerant (pLI) score=0), and biallelic loss-of-function variants have not been reported in apparently healthy individuals. This indicates that patients with POLE-associated diseases require residual POLE activity. A deep intronic mutation, which shows the leaky expression of a normal product,6 7 meets this criterion. However, it is sometimes difficult to identify pathogenic deep intron mutations using whole-exome sequencing alone. SpliceAI,14 an in silico splicing prediction tool, is useful to predict the impact on splicing, but could not predict the splicing of c.1226+234G>A. Thus, we need to consider cDNA analysis including RT-PCR as we performed in this study when IMAGE-I syndrome is suspected.
Our study suggests that all three missense and small indel mutations, c.711_713del, c.2593T>G and c.3019G>C, reduced localisation in the nucleus. POLE must maintain substantial nuclear localisation to replicate DNA. Other DNA polymerases, such as DNA polymerase alpha (POLA) and DNA polymerase delta (POLD), are imported to the nucleus to bind to their subunits. POLA and POLD are essential eukaryotic polymerases that complex with four accessory subunits similar to POLE. Research on a temperature-sensitive murine cell lines (tsFT20)15 and Drosophila melanogaster 16 have shown that POLA2 (DNA polymerase alpha subunit B) and POL32 (DNA polymerase delta subunit 3) are required for nuclear localisation of POLA, respectively. However, the three mutations in the N-terminal region or in catalytic polymerase domain do not alter the interaction with the three other subunits of the POLE complex (POLE2, POLE3 and POLE4), which are mediated via the non-catalytic C-terminal region.17 In fact, our study revealed that nuclear localisation was recovered by proteasome inhibition, suggesting that nuclear import of the mutants was not impaired. Previous reports have shown that mutant POLA proteins are degraded by proteasome-dependent degradation in the nucleus.15 Thus, the quality of DNA polymerases that play essential roles in cell proliferation may be maintained via a common mechanism. We observed that the aberrant polymerase was degraded in the nucleus; however, further analysis is required to elucidate how the mutation is recognised, which leads to its degradation in the nucleus.
Regarding delayed cell cycle progression and replication stress, it is noteworthy that both patients had complicated DLBCL. The relationship between POLE-associated syndrome and malignant diseases is often explained from two points of view. One is the somatic or germline mutation in the exonuclease domain,18 and the other is replication stress.19 In addition to polymerase activity, POLE possesses a proofreading function, and tumours having missense variants in the exonuclease domain show a hypermutated phenotype. c.711_713del and c.2593T>G were not present in the exonuclease domain, and we confirmed that each DLBCL tissue sample had low tumour mutation burden (data not shown). Replication stress is also involved in malignant diseases.20 In DNA replisome-associated diseases, DNA replication stress has been detected not only in POLE but also in cases of GINS1, MCM4 and POLD deficiency.21 However, only a few cases were complicated with malignancies, for example, osteosarcoma in GINS1 deficiency, and T-cell lymphoma and Hodgkin’s lymphoma in IMAGE-I syndrome (Nos. 5 and 15 in table 1). Additional mutations in these malignant samples have not been reported; thus, the involvement of replication stress remains unresolved. It is noteworthy that DLBCL in our two patients was associated with EBV infection. A patient with IMAGE-I syndrome complicated by EBV-associated haemophagocytic lymphohistiocytosis treated with allogeneic bone marrow transplantation has been reported previously (No. 1 in table 1). They probably could not efficiently control the EBV. Therefore, IMAGE-I syndrome should be considered cancer predisposition syndrome in terms of immunological disorders affecting immunosurveillance against tumours and EBV.
In conclusion, we identified two cases of IMAGE-I syndrome with biallelic POLE mutations. Both patients had common and novel deep intronic mutations. Our findings reinforce the hypothesis that a characteristic combination of a deep intronic mutation and loss-of-function mutation causes IMAGE-I syndrome in humans. Furthermore, aberrant POLE proteins showed severely diminished nuclear localisation due to proteasome-dependent degradation in the nucleus, providing us with new insights regarding the quality control of POLE functions.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by the ethics committee of Tohoku University School of Medicine (2019-1-561). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank the two patients who participated in this study and their families. We thank Dr S. Narumi of Department of Molecular Endocrinology, National Research Institute for Child Health and Development for research assistance; Y. Chiba and K. Itoh for technical assistance; and all colleagues in the Department of Paediatrics, Tohoku University Hospital for patient care. We also thank the Biomedical Research Core of the Tohoku University Graduate School of Medicine and the Biomedical Research Unit of Tohoku University Hospital.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors TNa, YS and AK designed the research, analysed the data and wrote the manuscript; TNa, KM, HN, TNi, MS and RF performed the research; KN, YA and SK supervised the project. YS acted as guarantor.
Funding This work was supported by grants from the Japanese Ministry of Health, Labour and Welfare (grant nos: 20FC1047 and 20FC1053), the Japan Agency for Medical Research and Development (grant nos: JP21gk01104h0003 and JP17ek0109151) and Grants-in-Aid from the Initiative on Rare and Undiagnosed Diseases (grant no: 21ek0109549s0901).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.