Article Text

Abstract
Rubinstein-Taybi syndrome (RTS) is a malformation syndrome characterised by facial abnormalities, broad thumbs, broad big toes, and mental retardation. In a subset of RTS patients, microdeletions, translocations, and inversions involving chromosome band 16p13.3 can be detected. We have previously shown that disruption of the human CREB binding protein (CREBBP orCBP) gene, either by these gross chromosomal rearrangements or by point mutations, leads to RTS. CBP is a large nuclear protein involved in transcription regulation, chromatin remodelling, and the integration of several different signal transduction pathways. Here we report diagnostic analysis of CBP in 194 RTS patients, divided into several subsets. In one case the mother is also suspect of having RTS. Analyses of the entireCBP gene by the protein truncation test showed 4/37 truncating mutations. Two point mutations, one 11 bp deletion, and one mutation affecting the splicing of the second exon were detected by subsequent sequencing. Screening theCBP gene for larger deletions, by using different cosmid probes in FISH, showed 14/171 microdeletions. Using five cosmid probes that contain the entire gene, we found 8/89 microdeletions of which 4/8 were 5′ or interstitial. This last subset of microdeletions would not have been detected using the commonly used 3′ probe RT1, showing the necessity of using all five probes.
- Rubinstein-Taybi syndrome (RTS)
- CREB binding protein (CBP/CREBBP)
- protein truncation test (PTT)
- microdeletion
Statistics from Altmetric.com
- Rubinstein-Taybi syndrome (RTS)
- CREB binding protein (CBP/CREBBP)
- protein truncation test (PTT)
- microdeletion
Rubinstein-Taybi syndrome (RTS) (MIM 180849), characterised by mental retardation, broad thumbs, broad big toes, and facial abnormalities, was described in 1963 by Rubinstein and Taybi.1 About three decades later, the first chromosomal translocations2 3 and microdeletions4associated with this syndrome were reported. In 1995, we cloned the gene encoding the human CREB binding protein (CREBBP, hereafter called CBP) (MIM 600140/GDB:437159) and found that mutations in this gene lead to RTS.5
CBP was first described in the mouse (Cbp) by Chrivia et al 6 in 1993 as a transcriptional coactivator. The protein was named for its interaction with the cyclic AMP regulated enhancer binding (CREB) protein. Although CBP and its homologue p300 were originally proposed to function as transcriptional coactivators by forming a physical bridge between the different components of the transcription machinery, recent evidence suggests that their potent histone acetyltransferase (HAT) activity “opens” the chromatin structure, allowing transcription factors access to the DNA. Furthermore, they are mediators of different signalling pathways and participants in basic cellular functions, such as DNA repair, cell growth, cell differentiation, apoptosis, and tumour suppression.7 Owing to the fact that CBP and p300 are central to multiple signal transduction pathways, thereby regulating the expression of many genes, it is difficult to relate this wide variety of functions to the characteristic manifestations of Rubinstein-Taybi syndrome.
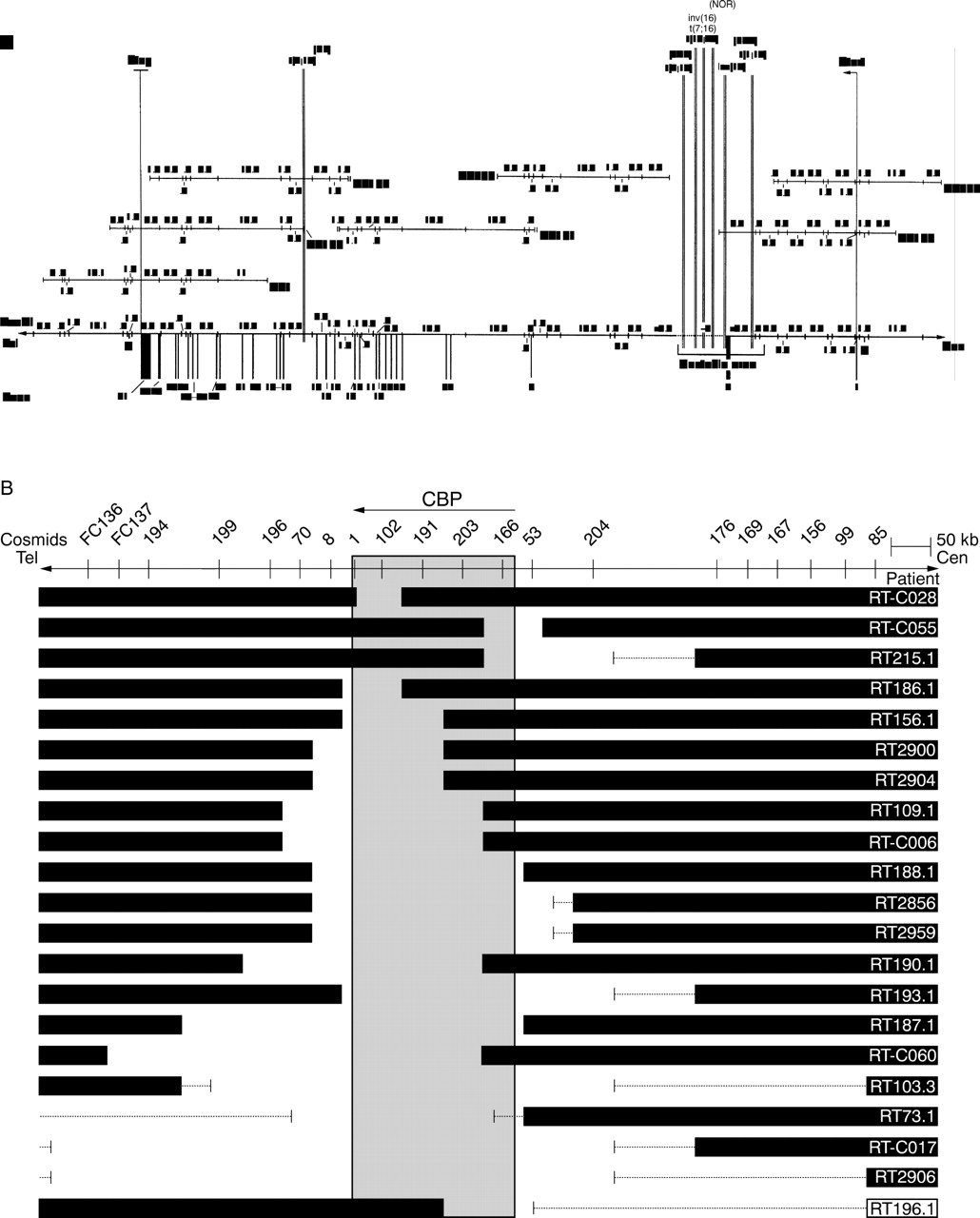
To date, all reports using FISH analysis to detect microdeletions in RTS patients have made use of the RT1 probe (D16S237). In 19/159 (12.0%) of these RTS cases, the FISH signal on one of the chromosomes 16 was lost.4 8-11 However, the RT1 cosmid probe only covers about 29 kb of the 3′ end of the CBPgene, whereas the transcribed part of the gene spans 146 kb of genomic DNA (fig 1A). We therefore extended the screening for microdeletions with four additional cosmids in order to cover the entire CBP coding region. In one of these 159 analysed cases a de novo translocation t(1;16) was detected.10 Five of the six other reported translocations and inversions (table 1) map to an area of approximately 13 kb at the 5′ end ofCBP (fig 1A). The breakpoints involved in acute myeloid leukaemia (AML) associated with the somatic translocation t(8;16) (p11;p13.3), in which a large part ofCBP is fused to an acetyl transferase gene called monocytic leukaemia zinc finger protein (MOZ) on 8p11,12-14 map to the same area.

Distribution of RTS gross chromosomal rearrangements. (A) Seven translocation and inversion breakpoints and their distribution over the CBP gene area, depicted in a minimal tiling path cosmid contig with a refined EcoRI/NotI restriction map. Numbers above and below the DNA line are EcoRI fragment sizes. N=NotI sites. Fragment sizes and exon-intron structure were established by comparing the genomic sequence of the CBP gene (GenBank Accession Nos AC 005564, 004495, 004509, 004651, and 004760) with that of the mRNA (GenBank Accession No U85962) (depicted without 5′ and 3′ UTR). (B) Schematic representation of 21 RTS microdeletions. When a microdeletion was found, flanking DNA up to 780 kb was screened by FISH with more cosmids in order to determine the size of the deletion. Black bars represent the undeleted DNA. Thin lines represent uncloned or untested areas.
Translocations and pericentric inversions reported in RTS
Previously we have shown that not only gross chromosomal rearrangements involving chromosome band 16p13.3 but also point mutations in the CBP gene lead to RTS.5 To obtain these results we used the protein truncation test (PTT), in which gene specific fragments are transcribed and translated in vitro in order to detect translational stops.15 Analyzing the first 1.2 kb ofCBP in a series of 16 patients with a clear clinical diagnosis of RTS, two were found to have a single base pair substitution resulting in a premature translation stop. These results suggested that the PTT was an appropriate method forCBP mutation detection in RTS. We then sequenced the entire CBPtranscript14 (GenBank Accession No U85962) and developed an additional series of primers covering all ofCBP. In this report we describe the PTT analyses of the entire 7.3 kb CBP coding region in eight overlapping fragments (varying in size from 0.6 to 1.9 kb) in an additional 35 RTS cases.
Methods
CLINICAL DIAGNOSIS AND PATIENTS
Rubinstein-Taybi syndrome is clinically defined by mental retardation, broad thumbs, broad big toes, growth retardation, and facial abnormalities. Important additional features are microcephaly, broadening of other fingers, radial deviation of the thumbs, malpositioned or crowded teeth with or without talon cusps, hypotonia, lax ligaments, stiff gait, hirsutism, cryptorchidism, glaucoma, recurrent respiratory infections, feeding problems, and keloid formation.1 16 The prevalence of RTS has been estimated to be 1 in 100 000 to 125 000 living newborns.17 All 194 patients in our study exhibited several features of the syndrome, establishing the clinical diagnosis. Patients originated from western Europe, Israel, and the United States and were referred to us for cytogenetic/molecular evaluation by clinical geneticists and paediatricians.
In one case the mother was also suspected of having RTS. At the age of 4 years the son had clear developmental delay with no speech (only sounds) and initial feeding problems. Furthermore, there were downward slanting palpebral fissures, long eyelashes, prominent nose, columella below the alae nasi, malpositioned ears with dysplastic helices, broad thumbs, somewhat broad toes, fetal pads, angulated penis, cryptorchidism, hirsutism, postnatal growth retardation, and a small head circumference. The karyotype was normal and no microdeletions could be detected (screened with five probes covering the entireCBP gene). We were not able to investigate the mother fully, but she had a prominent beaked nose with the collumella below the alae nasi, broad, short thumbs, which showed strikingly broad, short distal phalanges onx ray, broad big toes, long eyelashes, downward slanting palpebral fissures, and low intelligence. Her physical appearance is different from that of her two unaffected brothers, but she does resemble her son with RTS. She and her partner are non-consanguineous. The boy with RTS was their first born child and they later gave birth to a second healthy son.
Of the 194 patients in this report, 86 have previously been reported, 66 of them by us18 in a FISH study using the five cosmids covering CBP. The five microdeletions that were found were analysed in our present study for their deletion sizes and 14 non-microdeletion cases were checked for truncating mutations by PTT. Twelve non-microdeletion cases, which we also previously reported and included in a microdeletion detection study,14 were analysed for truncating mutations by PTT in this study. Four cases exhibiting RT1 microdeletions, reported earlier by Wallersteinet al,10 were analysed here for their deletion sizes. Three microdeletions and one translocation mentioned briefly in an earlier report5 were analysed in more detail. In total, 171 cases were investigated for microdeletions and translocations by FISH, and 37 cases for truncating mutations by PTT (table 2).
Molecular studies on patients with Rubinstein-Taybi syndrome
CYTOGENETIC ANALYSES
Almost all patients were karyotyped using standard cytogenetic methods followed by metaphase FISH analysis with one or more cosmid probes from the CBP gene area. The following cosmids were used: RT1, RT100, RT102, RT191, RT203, and RT166 (fig 1A and Giles et al 14). Together they cover the entire CBP gene, except for the ∼5 kb uncloned area between RT203 and RT166. For every cosmid, 30 metaphases per patient were examined for the presence of fluorescent dots on both chromosomes 16. At least 27 of the 30 metaphases must lack cosmid signal to be scored as deleted. In the beginning of our research we used cosmid RT1,4 which contains an insert of 51 kb. Because of instability this cosmid was replaced by RT100, which also covers the 3′ end of the CBP gene (fig 1A). Probes are made available online for diagnostic purposes at cost price by the Genome Technology Centre (GTC), Leiden. When a microdeletion was detected, further analyses with cosmid probes from the adjacent area(s) were performed in order to determine the size of the microdeletion (fig1B). One or two colour FISH was performed as described previously.19-21
RT-PCR, PTT, AND SEQUENCING
Total RNA was isolated from EBV transformed lymphoblastoid cell lines or peripheral blood and reverse transcribed with random hexamers. Primers used to amplify CBP cDNA fragments were selected with the help of Oligo 4.1 primer analysis software (National Biosciences) for optimal performance (table 3). PCR products were selected with a minimum overlap of 150 bp, necessary to have full coverage for the PTT; otherwise truncations of the artificially made protein fragments at the beginning or the end will be missed.
Protein truncation test PCR amplification of human CBP cDNA
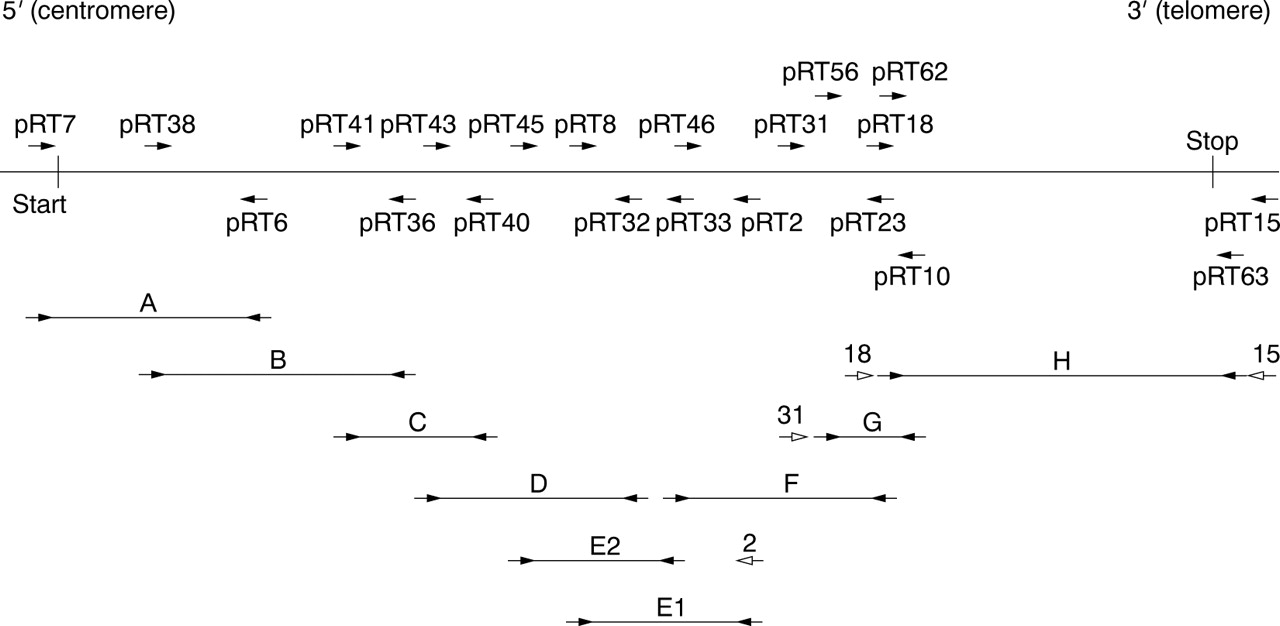
Expression levels of CBP are sufficient to perform a single RT-PCR for most fragments, directly using the PTT-primers (that is, a eukaryotic T7 promoter sequence and Kozak sequence added to a 5′ gene specific sequence and a second reversely orientated gene specific primer) on the cDNA pools (fragments A-D, E1, and F). For some patients, a second (booster) PCR with the same primer set had to be performed to improve yield. The PTT often requires the use of a primary PCR from which a booster PCR is performed using the PTT-primers in a nested or hemi-nested PCR reaction. In fragments E2, G, and H, the PTT primers were used in such a (hemi-) nested fashion (fig 2). In the case of pRT7, we used the endogenous translation initiation site and 15 bp of the 5′ UTR ofCBP instead of the standard eukaryotic in frame translation initiation sequence.
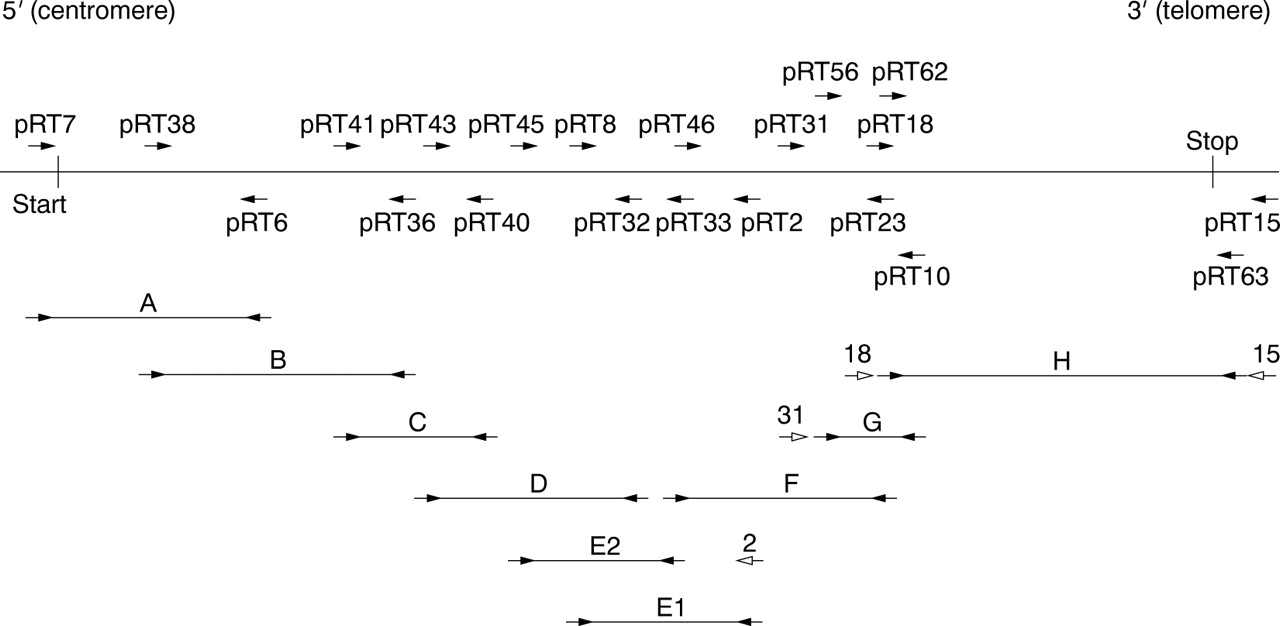
RT-PCR fragments for the protein truncation test (PTT) covering CBP. Arrowheads represent primers used. Open arrowheads represent primers used in primary PCRs for those fragments requiring both a primary and secondary (booster) PCR.
In all PCR reactions we used 10 pmol of each primer, 1.5 mmol/l MgCl2, 10 mmol/l Tris-HCl, pH 8.9, 50 mmol/l KCl, 0.01% gelatine, 10% glycerol, 0.2 mg/ml BSA, 0.2 mmol/l dGTP, 0.2 mmol/l dATP, 0.2 mmol/l dTTP, 0.2 mmol/l dCTP, and 3 U Gibco BRL recombinantTaq DNA polymerase. The buffer was stored in a 10 × concentration at −20°C without glycerol, MgCl2, or Taq DNA polymerase and was frozen at least once before use. Initial denaturing was performed for four minutes at 94°C, followed by 40 cycles of one minute denaturing at 94°C, one minute annealing at 57°C, 1.5 minutes extension at 72°C, and then final extension for five minutes at 72°C. A Hybaid Ltd OmniGene temperature cycler with Biozym thin wall 0.5 ml microcentrifuge tubes were used. The end volume of the reactions was 50 μl. Booster PCRs were performed in the same fashion by using 1 μl of the primary PCR product as template and only 30 PCR cycles. In the 1.9 kb fragment H, best results were obtained by extending the extension at 72°C to two minutes and by using 32 cycles in both primary and booster PCR. Since this fragment is located withinCBP's large final exon,14 this PCR was performed on 30-100 ng genomic DNA. All PCR products were analysed for yield and quality on standard 0.8% agarose gels stained with ethidium bromide.
In this study we performed the protein truncation test as described by Roest et al.15 Sequencing was performed with a USB/Amersham Sequenase PCR Product Sequencing Kit according to the manufacturer's recommendations or by standard dideoxy terminating chemistry on a Perkin Elmer ABI automated sequencer using the Cycle Sequencing Ready Reaction Kit (Perkin Elmer, Cat No 430349).
DNA EXTRACTION AND SOUTHERN BLOT HYBRIDISATION
Genomic DNA was isolated directly from peripheral blood or EBV transformed lymphoblastoid cell lines. In the RTS case where the mother is also suspected of having RTS, genomic DNA was digested withBamHI, BglII,EcoRI, EcoRV,HindIII,PstI, SfiI,StuI, and StyI restriction endonucleases in One-Phor-All buffer PLUS (Pharmacia). DNA was electrophoresed on standard agarose gels and transferred to Hybond N+ (Amersham) nylon membranes according to the manufacturer's recommendations. Hybridisation probes were 32P radiolabelled using random hexamers (Amersham), and repetitive elements were blocked by competition with 120 μg of sonicated human placenta DNA for two hours. Membranes were hybridised overnight at 68°C, washed stringently, and autoradiographed overnight.
Results
KARYOTYPING
In one patient of Dutch origin, karyotyping showed a translocation t(2;16)(q36.3;p13.3). In a metaphase FISH experiment with probe RT1 the translocation was confirmed; all the signal of this chromosome 16 probe is translocated to the derivative chromosome 2 (not shown). Parental chromosomes were normal.
MICRODELETION DETECTION BY FISH
Of the 171 patients who were tested for microdeletions, 14 (8.2%) lacked one or more cosmid signals. For 11 patients, sufficient material was available for testing flanking cosmids in order to determine the sizes of their deletions. In addition, four RT1 microdeletion cases reported earlier by Wallerstein et al 10 were analysed for their deletion size. To show deletion size distribution we included six earlier reported cases as well,5 8 resulting in a total of 21 cases depicted in fig1B. A total of 17/21 showed at least a RT1/RT100 deletion, which corresponds to the 3′ end of the gene. Interestingly, not all deletions could be detected with the RT1/RT100 probe. In the four (4/21) remaining cases, the 3′ end of the CBP gene was not affected and showed 5′ deletions (patients RT-C055, RT196.1, and RT215.1) or interstitial deletions (patient RT-C028) (fig 1B). Microdeletion sizes varied from <50 kb (patient RT-C028) to >650 kb (patient RT2906).
CBP TRUNCATION DETECTION BY PTT
To search for truncating mutations we performed the protein truncation test on the entire CBP coding region of 37 cases in eight overlapping PTT fragments. Four (4/37) mutations were found. Two had been previously reported5; in both cases a codon for glutamine was changed into a stop codon by a C→T substitution. Mutations occurred at base pair positions 406 and 1069, which are positioned in PTT fragment A (fig 2). In a third case, PTT followed by sequence analyses showed a deletion of 11 bp (del4080-4090) leading to a frameshift and a premature stop 22 amino acids further downstream (patient RT113.1/PTT fragment E2). In fig 3the truncated protein and the sequence analysis is shown. No significant differences were found between the clinical features of these three patients with a truncating mutation and those with a microdeletion.

Protein truncating mutation of patient RT113.1. (A) PTT of fragment E2 (CBP position 3284-4640) of patient RT113.1 (lane 2) and 11 other RTS patients. The truncated protein is indicated by an arrowhead. (B) Standard dideoxy terminating chemistry on a Perkin Elmer ABI automated sequencer with pRT55 used as primer. The del4080-4090 mutation is indicated by the box. (C) The del4080-4090 mutation leads to a frameshift and a premature stop (TGA in bold) 22 amino acids further downstream.
A fourth aberration was detected on the agarose gel before we performed in vitro transcription/translation. In this case the PCR product of fragment A (with primers in exons 1 and 4) showed a very faint 1.2 kb normal product and an abundant aberrant fragment of 0.5 kb (fig 4, lanes 7 and 8). Sequencing of this RT-PCR product showed a 713 bp deletion in the mRNA which starts at nucleotide position 86 and ends at nucleotide position 798, resulting in a frameshift and translational stop 13 amino acids further downstream. The cDNA deletion exactly corresponds to the second exon ofCBP 14 (fig 1A). Because the patient's mother was also suspected of having RTS, we performed RT-PCR on her DNA as well. The mother showed similar results to her son, although the intensity ratio between the two bands was approximately equal (fig 4, lanes 3 and 4). Surprisingly, the 0.5 kb PCR product can also be seen in the father and other controls (n=40), but is markedly less pronounced (fig 4, lanes 2, 5, 6, and 9). We isolated the 0.5 kb fragment of the father and sequence analysis confirmed that this product also lacks exon 2. These results suggest an error in splicing. Sequence analysis of the splice site and the concomitant branch site, however, did not show any changes. By Southern analysis with several restriction enzymes (see Methods) we could not detect any large deletions at the DNA level (not shown).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
RT-PCR analysis of a two generation RTS case. pRT7 and pRT26 RT-PCR products (CBP position –15-1126) electrophoresed on an ethidium bromide stained agarose gel. Lane 1, 100 bp marker; lane 2, one sample of a control; lanes 3 and 4, two samples of the mother; lanes 5 and 6, two samples of the father; lanes 7 and 8, two samples of the child with RTS; lane 9, one sample of the healthy brother. The second sample of the mother, father, and child were obtained by separate mRNA isolations. The normal product is 1.2 kb in size; the alternatively spliced product is 0.5 kb.
Discussion
Adding the data presented here to those previously reported, we can conclude that translocations and inversions form the minority ofCBP mutations in RTS. We only found one translocation in our set of 171 patients. Micodeletions occur more frequently. In a first series of 24 patients with the clinical features of Rubinstein-Taybi syndrome, we detected six microdeletions (25%).4 In our present study of 171 patients, we now detect 14 microdeletions (8.2%). The difference in microdeletion frequency between the initial study and the present one may be for several reasons. For instance, the 171 samples were sent to us by many colleagues from different countries. There may have been slight differences in the diagnostic criteria used by this large group of clinicians. The percentage of 25% microdeletions found in the first series may have been an overestimation owing to small numbers. The true 16p microdeletion frequency in RTS appears to be around 10%.
As seen in fig 1B, all microdeletions cause a complete or partial deletion of the CBP gene. Partial deletions of both the 3′ and the 5′ end of the gene have been identified. Because many of the microdeletion cases were initially investigated for the presence of RT1/RT100 signal, data bias could occur. In order to exclude a 3′ selection bias, 89 of our 171 patients were tested for microdeletions with all five cosmids simultaneously. In this subset, 8/89 (9.0%) microdeletions were found. Four of these eight (50%) were not deleted for RT1/RT100 and would therefore have been missed using standard RT1/RT100 screening methods. These data stress the importance of using all five cosmids covering the entireCBP gene to detect all microdeletions in patients with a clinical picture of RTS.
No other genes besides CBP have been mapped to the area shown in fig 1B. At the 5′ side ofCBP none of the 18 tested microdeletions extended beyond RT176 (three cases were excluded because they could not be tested in this region). This would imply that other known centromeric genes such as phosphomannomutase 2 (PMM2) (MIM 601785), involved in carbohydrate deficient glycoprotein syndrome type I (CDG1) (MIM 212065),22 are not deleted in these RTS microdeletions. On the 3′ side, however, some of the microdeletions extend towards the chromosomal region which is known to contain the gene for familial Mediterranean fever (MEFV) (MIM 249100).14 23 Because MEFV is an autosomal recessive disease, we do not expect symptoms in RTS patients that potentially have deletions within theMEFV gene area. If, however, the deletions extend into the PKD1 andTSC2 genes, cysts in the kidneys and stigmata of tuberous sclerosis could be expected. These features have not been described in RTS patients. Since deletions spanning this entire region have not been reported in any subject, with the exception of a ring(16) chromosome, which was later found to harbour the relevant part of 16p on the short arm of chromosome 1, it was postulated earlier that the region between CBP andPKD1 may contain one or more genes of which a diploid dose is essential for survival.4
The fact that CBP is prone to translocations, inversions, and deletions suggests the presence of elements conferring genomic instability. As would be expected in such a case, rearrangements affecting the CBP gene are not evenly distributed. In fact, six out of seven studied RTS translocations exhibit breakpoints within the same 13 kb breakpoint region of intron 2,14 as well as six of the microdeletion breakpoints depicted in fig 1B. The breakpoints of the AML associated somatic translocation t(8;16)(p11;p13.3) map to the same area.13 14 Interestingly, the 13 kb breakpoint interval contains a 5 kb region that has proven unclonable in multiple attempts made by several groups to date.12 14 Sequencing this breakpoint cluster region will hopefully lead to an explanation for the instability of this region.
To examine more subtle mutations at the transcript level we turned to the PTT. In a previous study we reported protein truncating mutations in two patients by using the PTT on the first 1.2 kb of theCBP transcript.5 These early results indicated that PTT could be a quick method to screen forCBP mutations in RTS patients. To estimate the number of truncating mutations in a larger series of patients we screened the entire CBP transcript in 35 patients. Only two additional mutations were found, adding up to 4/37 (10.8%) truncating mutations found by RT-PCR or PTT or both. This low number of truncating mutations is confirmed by a pilot study, in which we performed single stranded conformation polymorphism analysis (SSCA) of the last CBP exon (29% of the transcript) on the DNA of 111 RTS patients (partly overlapping the 194 patients reported here). Three (2.7%) nonsense mutations were found (unpublished data). Taking all our data into account it becomes questionable whether the effort and costs are worth the yield and if the PTT technique is suitable for a regular diagnostic setup for RTS. If protein truncating mutations could be detected directly on western blots, high costs and tedious multiple PTT tests could be avoided.
If fertility is not impaired one could expect a 50% recurrence risk for any haploinsufficiency disease. Two families have been described in which a mother and child both had RTS.24 25 In both families, however, the children clearly had more pronounced dysmorphic features and were mentally more retarded than their mother. RTS diagnosis in the mothers would have been difficult without the more pronounced phenotype of their children. Parents of RTS children should always be evaluated carefully. These two cases and the one we present in this paper illustrate once more the variability of the syndrome. Recent mouseCbp/p300 knock out experiments indeed show that the genetic background is a major feature in phenotype and survival.26 27 In the mother and child presented here, we could not determine the exact genomic mutational mechanism. At the RT-PCR level, the mother and child both show alternative splicing of Cbp. The fact that RNA of lymphoblastoid cells of normal subjects show similar alternatively spliced products (but in a much lower dose) and no genomic deletions could be detected in the mother and child indicate that a mutation could be present in one of the splice regulatory mechanisms. However, mutations in the branch site or the splice acceptor site of the skipped second exon were excluded by sequence analysis. Alternatively, post-transcriptional processing can also be influenced by intronic repeats,28 silent exonic sequence variation,29 30 and other unknown mechanisms. TheCBP introns 1 and 2 and exon 2 should be analysed to determine any sequence variance in this two generation case as compared to a series of controls. Introns 1 and 2 are respectively 28.8 kb and ∼33.3 kb in size and include the unstable region (fig1A), which makes analysis of the flanking introns difficult. Whether the 43 amino acid protein product of the alternatively splicedCBP is stable and whether it has any function is unclear at this moment. The first 102 amino acids of CBP contain a nuclear hormone receptor interacting domain.31 32 Thus the alternative product may still bind the nuclear hormone receptors resulting in a dominant negative effect.
The fact that we only found ∼20% CBPmutations (either by karyotyping and FISH or by PTT) in a large set of RTS patients and that RTS is a syndrome with a highly variable phenotype suggests that not only loss of one functional copy ofCBP but also that mutations in other genes could play a role in the aetiology of RTS. Other mutational mechanisms could include CBP promoter (or other regulatory element) mutations, CBP missense mutations (71% of the gene still has to be investigated), intronic mutations within the uncloned 5 kb area that affect transcription, silent mutations in exons, or homologous intrachromosomal recombination between repeated sequences such as seen in haemophilia A (MIM 306700).33 Other genes involved could beCBP homologues, such as the closely relatedp300 on chromosome band 22q1334 35 or genes that encode proteins that participate in the different signalling pathways in which CBP is involved. From mouse knock out experiments it is known that losing one allele ofp300 or Cbp leads to major health problems.26 27 36 HeterozygousCbp animals show skull and rib cage deformities, and heterozygous mutants for eithercbp or p300 have a higher chance of dying during pregnancy (up to 50% depending on the background). Losing two alleles of eithercbp or p300 leads to intrauterine death. Interestingly, the compound heterozygous mouse (Cbp +/-, p300+/-) exhibits the same phenotype as either thecbp or p300homozygous mutant mice.27 One could postulate a “triploinsufficiency model” in which losing one of the fourCBP/p300 alleles leads to major health problems and losing two alleles leads to early death. However, in contrast to cbphemizygous mice, p300 hemizygotes do not show RTS-like features. This makes p300 less appropriate as a candidate gene for RTS. Furthermore, no RTS patients have been described with translocations or inversions involving chromosome band 22q13.
Further insights into how mutations in CBPcould lead to the RTS phenotype might come from other malformation syndromes, especially Greig's cephalosyndactyly syndrome and Saethre-Chotzen syndrome. Greig's syndrome, which is similar to RTS in limb and craniofacial dysmorphisms but does not include mental retardation, is caused by mutations in theGli3 gene.37 Gli3 is highly homologous to theDrosphila gene Cubitus interuptus (CI), a transcription factor in the Hedgehog signalling pathway and a member of the Gli signalling pathway. In studies using mutated Drosophila CBP (dCBP), it was determined that dCBP functions as a cofactor for CI.38 The similarity between Saethre-Chotzen syndrome and RTS is even more striking, illustrated by the publication of Lowryet al 39 in which a Saethre-Chotzen patient appeared to have been incorrectly diagnosed with RTS. Saethre-Chotzen syndrome is caused by haploinsufficiency of the human TWIST gene product.40The previously mentioned dCBP mutants were observed to affect the Dorsal dependent expression of the Twist gene.41Because the Dorsal protein recruitsdCBP to initiate transcription ofTwist, it seems likely that haploinsufficiency of CBP could influence the expression ofTwist. Low expression of the Twist protein, either by mutations in Twist itself or by low levels of a cofactor involved in its expression (such asDorsal), could lead to the clinical overlap between Saethre-Chotzen and Rubinstein-Taybi syndromes. Moreover,Twist was recently shown to bind to the HAT domain of p300 itself and thereby directly regulates its HAT activity.42
Microdeletions of and truncating mutations inCBP account for ∼20% of the mutations in RTS. CBP interacts with multiple proteins and participates in several signalling pathways. The remaining mutations responsible for RTS could therefore be hidden in a large number of genes.
Acknowledgments
The authors wish to thank Annemieke Rus, Sander Kneppers, and Norman Doggett for their technical assistance or sharing unpublished data. This work was supported by the Dutch Cancer Society (IKW92-24), the Dutch Organization for Scientific Research (NWO 901-04-124), and grants from the European Community (CT93-0040). Accession numbers and URLs for data in this article are as follows: Genome Technology Centre (GTC) - Leiden,http://ruly70.medfac.leidenuniv./∼gtc/vecprobe.html#RT (for cosmid probes made available at cost price); Online Mendalian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for MIM numbers); GenBank, http://www.ncbi.nlm.nih.gov/Entrez/nucleotide.html (for nucleotide sequences).