Article Text

Abstract
Classical neonatal diabetes mellitus is defined as hyperglycaemia occurring within the first six weeks of life in term infants. Cerebellar agenesis is rare. We report three cases of neonatal diabetes mellitus, cerebellar hypoplasia/agenesis, and dysmorphism occurring within a highly consanguineous family. This constellation of abnormalities has not previously been described. Two of these cases are sisters and the third case is a female first cousin. The pattern of inheritance suggests this is a previously undescribed autosomal recessive disorder. Prenatal diagnosis of the condition in this family was possible by demonstration of the absence of the cerebellum and severe IUGR.
- cerebellar agenesis/hypoplasia
- neonatal diabetes mellitus
- dysmorphic features
- autosomal recessive
Statistics from Altmetric.com
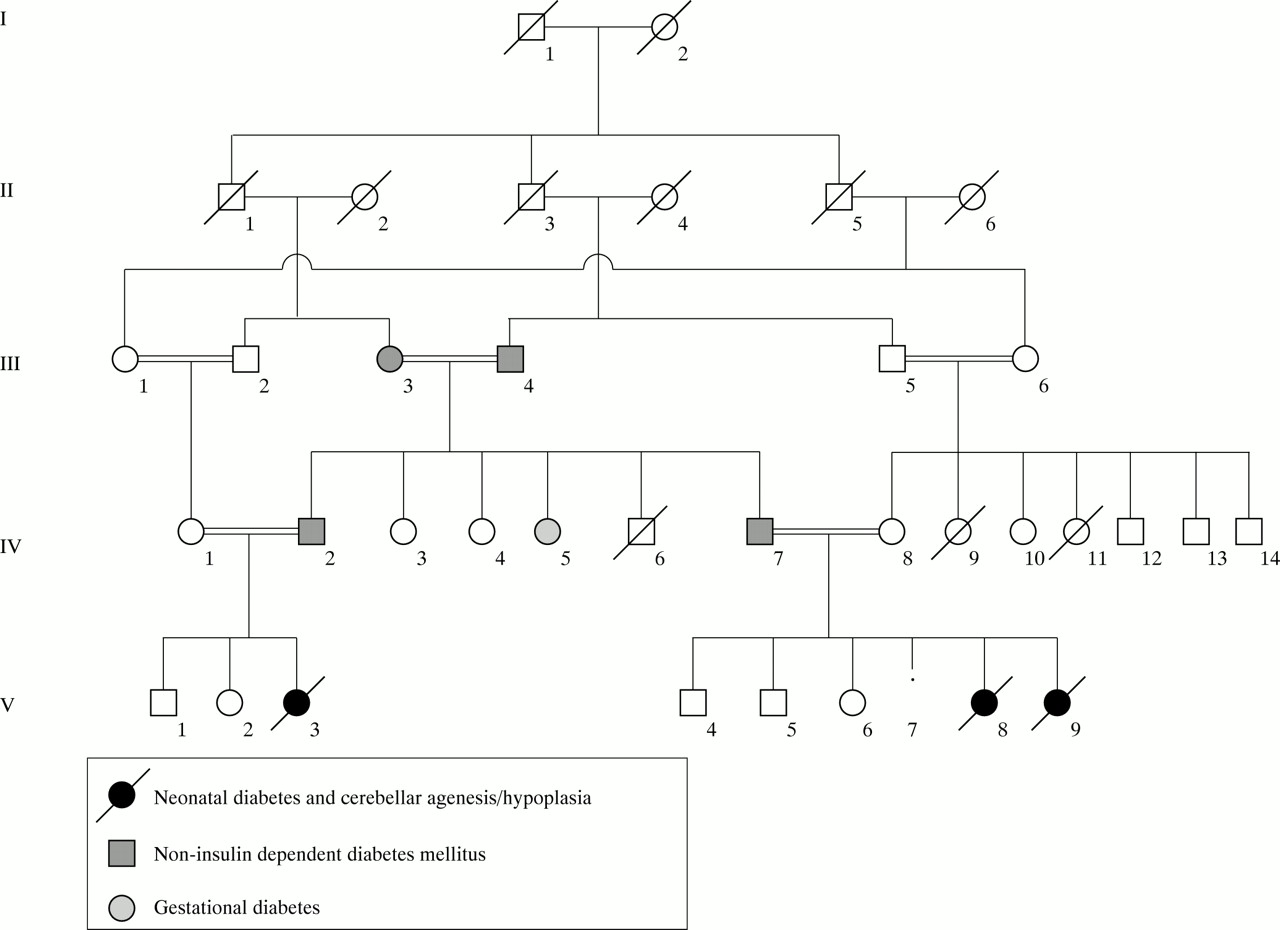
Classical neonatal diabetes mellitus (NDM) is defined as hyperglycaemia occurring within the first six weeks of life in term infants.1 A recent survey by the British Paediatric Association Surveillance Unit (BPASU) has established an incidence of 1 in 400 000 live births2; in a similar German study the incidence was 1 in 500 000 neonates.3 Complete cerebellar agenesis is rare and there are only a few published reports.4-6 We describe two female sibs and a female first cousin in a highly consanguineous family with cerebellar hypoplasia/agenesis and neonatal diabetes mellitus. The clinical features are shown in table 1. This association has not previously been reported.
Clinical features
Case reports
CASE 1
A female infant (V.8), the fourth child of consanguineous, Asian, immigrant parents both aged 34, was born two weeks post-term. Her parents and both maternal and paternal grandparents were first cousins (fig 1). There was a strong family history of non-insulin dependent diabetes mellitus (NIDDM) in the absence of obesity. Her father developed NIDDM at the age of 37, the paternal grandmother at the age of 38, the paternal grandfather at the age of 74, and a paternal uncle (IV.2, the father of case 3, V.3) at the age of 34. Her paternal aunt (IV.5) had gestational diabetes. A physically normal paternal aunt (IV.3) was mentally subnormal. A maternal aunt (IV.9) died at 4 weeks (cause unknown) and a maternal cousin was stillborn. The mother was gravida 5, para 4. Her first pregnancy produced a small, growth retarded male infant after which she had a normal female and a normal male infant. The fourth pregnancy resulted in a miscarriage at 8-10 weeks’ gestation (sex unknown).
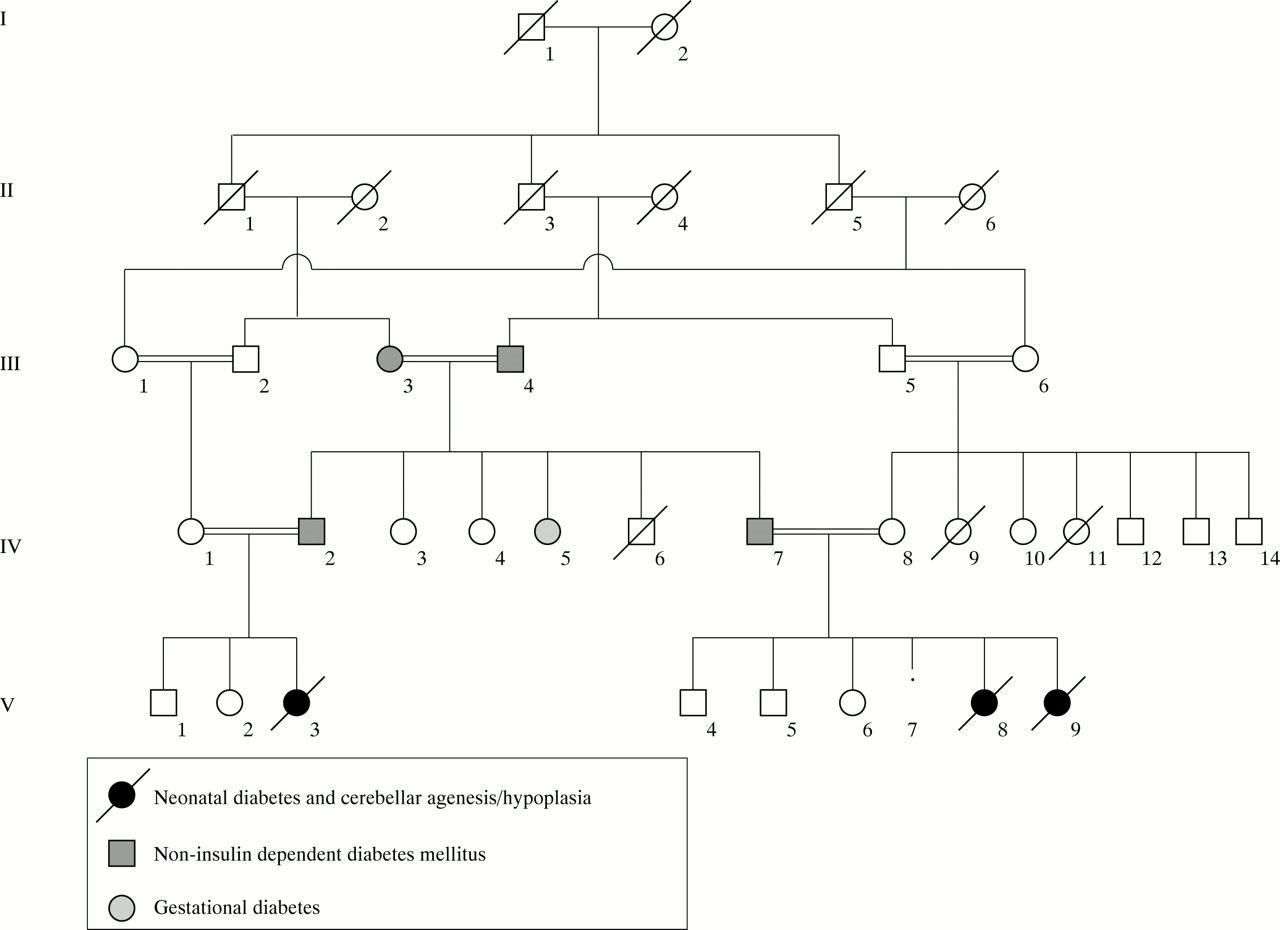
Family pedigree.
This infant had severe intrauterine growth retardation (IUGR), weighed 1540 g (<0.4th centile), and had a head circumference of 30 cm (<0.4th centile). She had a beaked nose and low set, dysplastic ears. She had very little subcutaneous fat, bilateral talipes equinovarus, and joint stiffness, especially of the hips. She was not unusually hirsute. Her respiratory pattern was irregular, requiring ventilation. She was noted to have abnormal movements from the first day. A subsequent interictal EEG showed increased cerebral excitability. As her abnormal movements persisted she was treated with intravenous phenytoin with good response. A diagnosis of NDM was based on raised blood glucose; the highest level recorded on the first day was 40 mmol/l (normal range 2.0-5.5 mmol/l). Despite a continuous insulin infusion ranging from 0.05-0.1 U/kg/h, adequate glycaemic control proved to be difficult.
Her progress was unsatisfactory. She required intermittent ventilatory support for a very irregular respiratory pattern. Despite administration of isophane insulin subcutaneously at a dose of 1.3 U/kg/day, glycaemic control remained poor, punctuated by sudden and severe hypoglycaemia. Although her oral feeds were increased to a maximum of 200 ml/kg/day, she failed to gain weight and at 3 months of age her weight was 1.2 kg, 300 g below her birth weight. She developed total parenteral nutrition (TPN) related jaundice and had multiple transfusions for anaemia.
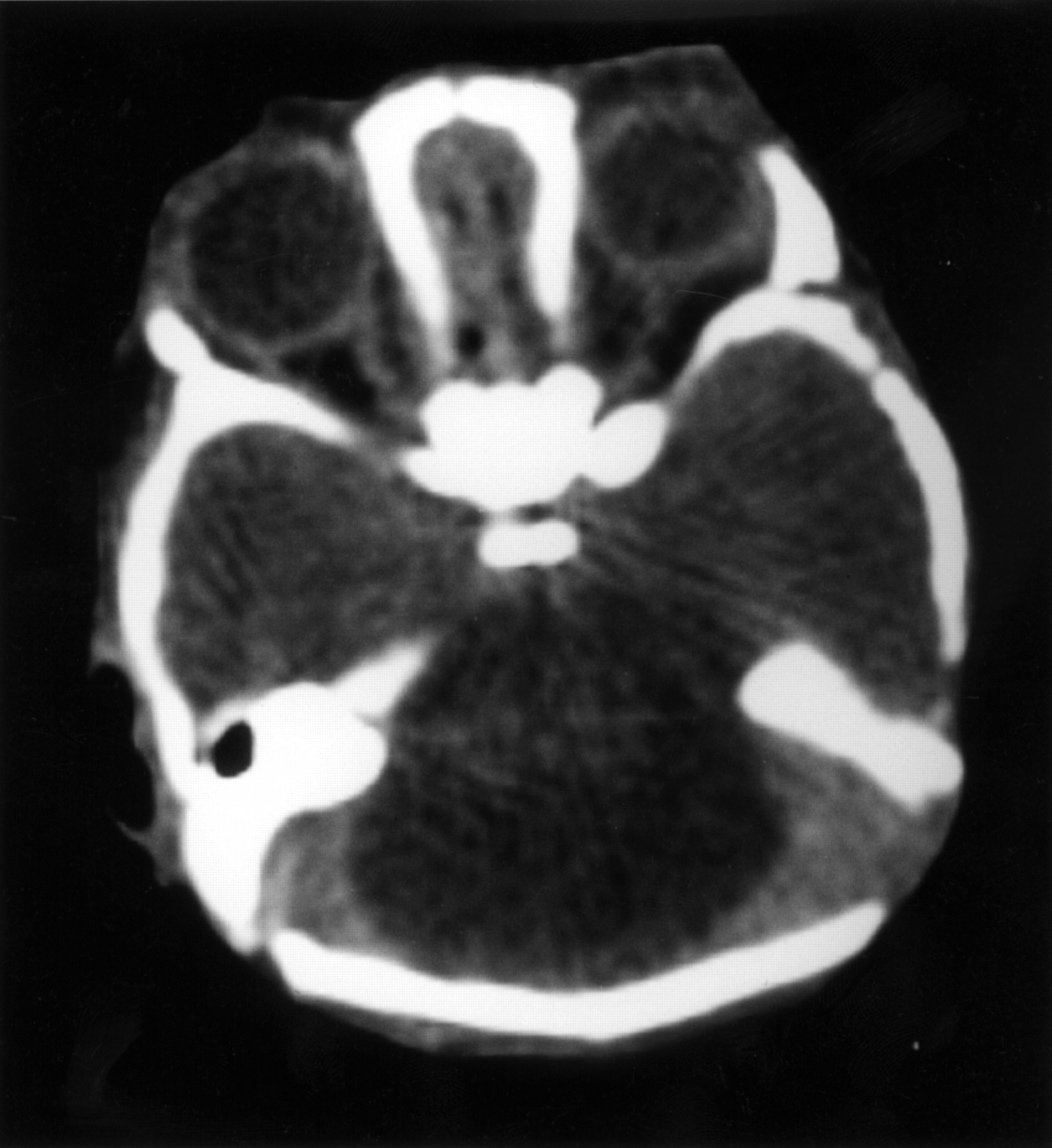
C peptide levels were low (88 pmol/l), with very low to undetectable levels of insulin (<25 pmol/l) in the presence of hyperglycaemia, and insulin autoantibodies were negative. She also had conjugated hyperbilirubinaemia (maximum level 132 mmol/l, normal 1-17 mmol/l) which improved after stopping TPN. Laboratory investigations are shown in table 2. Her cranial CT scan showed marked cerebellar hypoplasia (fig 2) but no other abnormalities were noted. She had a normal female karyotype and uniparental disomy of chromosome 6 (UPD6) was excluded. DNA analysis for the IPF1 gene mutation, Pro63fsdelC, was negative. She did not show any developmental progress and died at the age of 3½ months from presumed sepsis. Her parents declined permission for necropsy.
Investigations
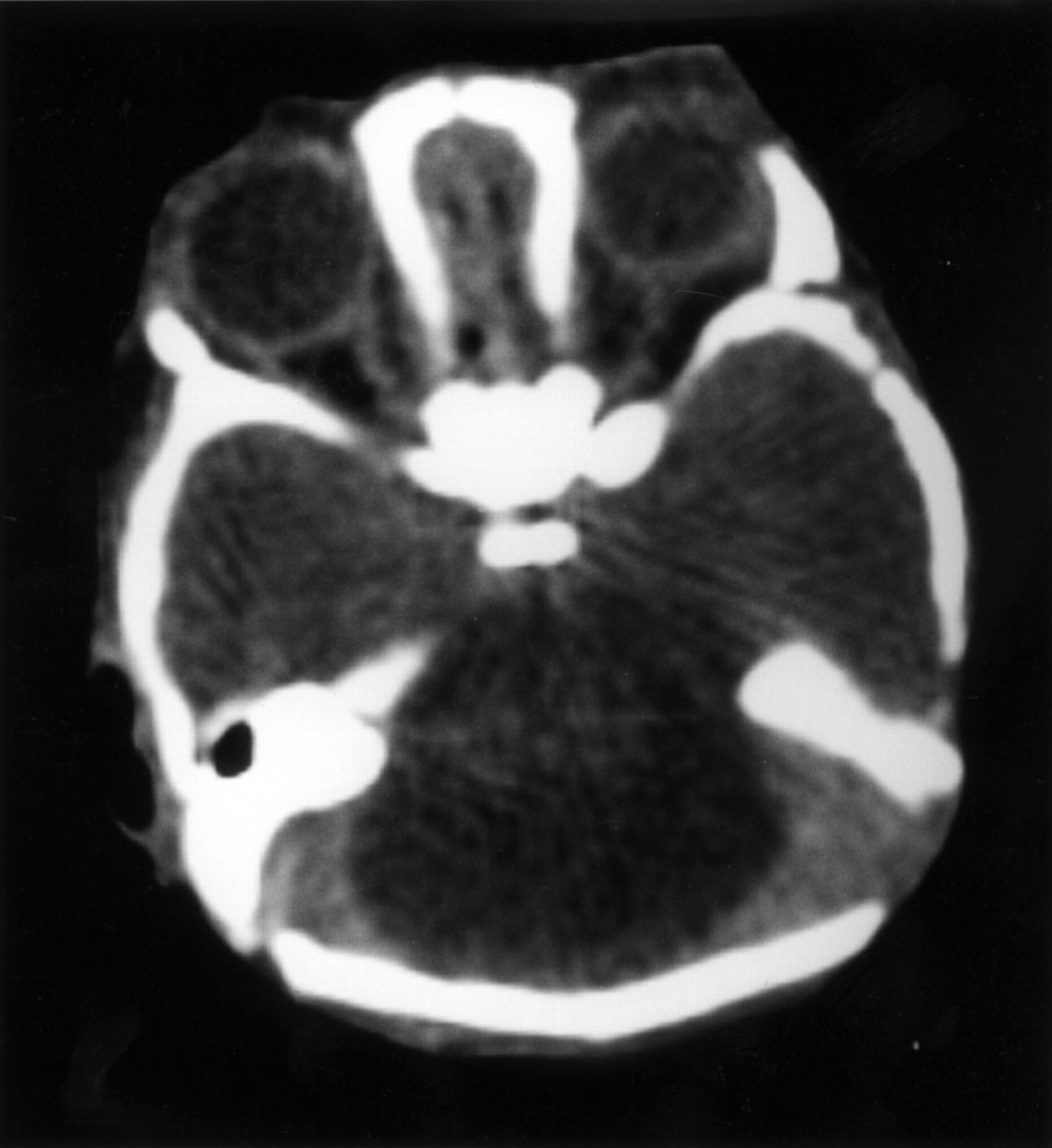
The axial cranial CT scan of case 1, showing marked cerebellar hypoplasia.
CASE 2
A further pregnancy followed a year after the death of the first patient. A fetal anomaly scan at 23 weeks’ gestation showed absence of the cerebellum, increased liquor, and severe IUGR. The parents declined the option of termination of pregnancy. A female infant (V.9) was born at 37 weeks’ gestation by vaginal delivery, weighing l390 g (<0.4th centile), with a head circumference of 28.9 cm (<0.4th centile). She was also dysmorphic with a wizened triangular face, low set, malformed ears, and very little subcutaneous fat (fig 3). She had bilateral talipes equinovarus and joint stiffness. This infant, like her previous sib, had an irregular respiratory pattern, with episodes of apnoea. In view of the prognosis and following discussion with the parents, her carbon dioxide retention was not treated with ventilation. She was started on a caffeine infusion and given oxygen via nasal cannula.

Dysmorphic facial features of case 2. Note that all three cases had similar dysmorphic features.
Blood glucose was 24.1 mmol/l on the first day of life and 36.9 mmol/l on the second day. She was started on intravenous insulin at 0.025-0.1 U/kg/h and then subcutaneous isophane insulin was administered at a dose of 0.6 U/kg/day. As in case 1, blood glucose control remained erratic, punctuated by sudden and severe hypoglycaemia. An MRI scan of her brain confirmed the diagnosis of cerebellar agenesis. There was also some slight delay in the maturation of myelin (fig 4A, B). Ophthalmological examination showed very pale and small discs with optic nerve hypoplasia.

{kind=link}
{kind=link}
{kind=link}
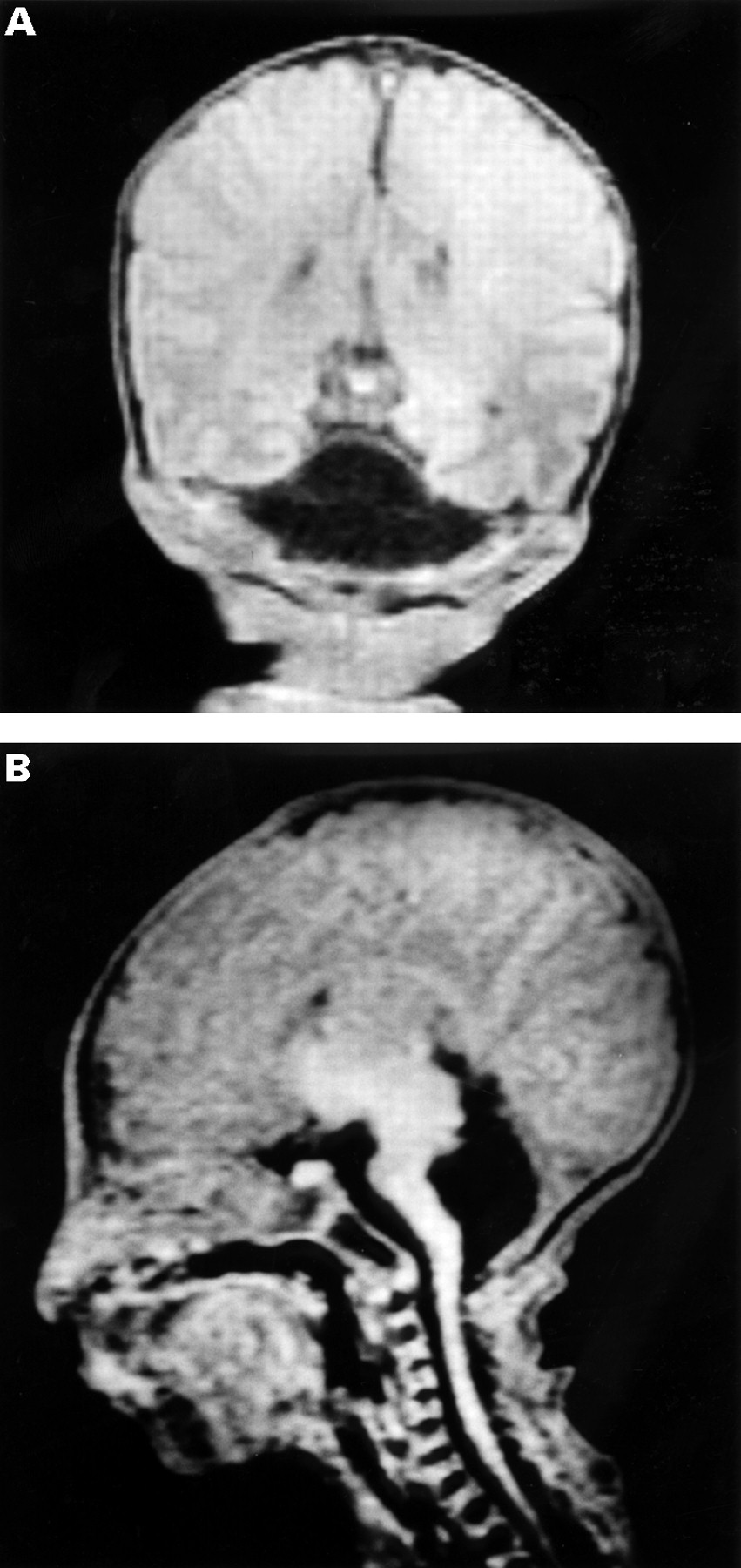{kind=link}
Cranial MRI scan of case 2. Coronal T1 weighted MRI (A) and sagittal T1 weighted MRI (B) at 3 days of age showing absence of the cerebellum.
C peptide level was low (<165 pmol/l) with a low insulin (2.7 mU/l), in the presence of hyperglycaemia (32.1 mmol/l). (Assays of insulin level were different in cases 1 and 2. The assay for case 1, performed at the Royal Surrey County Hospital, Guildford in 1995, was an “in house” immunoradioassay (RIA). The assay for case 2, performed at the Hammersmith Hospital in 1997, was an “in house” immunoradiometric assay (IRMA).) Serum immunoreactive trypsinogen (IRT) was very low at 3 mg/l. (Serum IRT result obtained from Peterborough District Hospital; an IRT of <10 mg/l suggests pancreatic exocrine insufficiency (normal neonatal reference range: mean (2 SD)=11-176 mg/l).) Thyroid function was normal, the IgE was <1 kU/l (0-6 kU/l), and UPD6 was again excluded. Ultrasound of her abdomen showed normal kidneys, liver, and common bile duct, but the pancreas was obscured by bowel gas. Chromosomal analysis showed a normal female karyotype. Carbohydrate deficient glycoprotein syndrome (CDGS) was excluded as no atypical transferrin glycoforms were detected in the blood. There was no evidence of the 3243 mitochondrial mutation associated with non-insulin dependent diabetes mellitus.
This infant’s progress remained poor with slow weight gain, recurrent anaemia requiring blood transfusions, and a compensated respiratory acidosis. She died at the age of 6 weeks from presumed sepsis. Necropsy was again refused.
CASE 3
Case 3 (V.3) was a female first cousin of the above sibs. The 33 year old father was the brother of the father of cases 1 and 2, and the 26 year old mother was their first cousin (fig 1). They had a son and daughter who were well but who had been growth retarded in pregnancy.
Case 3 was born at 33+ weeks’ gestation by emergency caesarean section for fetal distress, following a pregnancy complicated by oligohydramnios. She weighed 1200 g (<0.4th centile), had a head circumference of 27.8 cm (<2nd centile), and was dysmorphic with a small chin, triangular face, and low set ears. There was overlapping of the fingers of both hands and she had severe bilateral talipes equinovarus. She was intubated and ventilated for three minutes for poor respiratory effort. After extubation she was noted to have rapid, shallow breathing followed by brief periods of apnoea. As blood glucose on the first day of life was 28.8 mmol/l, she was started on intravenous insulin 0.0125-0.25 U/kg/h. Her postnatal course was punctuated by episodes of clinical sepsis including staphylococcal septicaemia. She had recurrent abdominal distension, was intolerant of feeds, and she failed to gain weight. Her insulin requirements varied but declined progressively. She required several blood transfusions for anaemia. Ophthalmological examination showed small pale discs with surrounding pigment.
A cranial CT scan showed cerebellar agenesis. An abdominal ultrasound scan confirmed the presence of some pancreatic tissue, a finding corroborated by the presence of normal faecal tryptic activity. C peptide was again low (<165 pmol/l). She had a normal female karyotype and UPD6 was again excluded. This infant died on day 55 from presumed sepsis. Consent for necropsy was not given.
Discussion
Typically NDM is associated with severe IUGR. This might be related to the apparent failure of insulin secretion in utero as insulin is a known growth factor. NDM differs from classical type 1 diabetes of childhood as the aetiology is probably not autoimmune in nature and its course is highly variable. Some patients have permanent diabetes, but others have transient or lasting remissions.2 3
NDM may occur in isolation or may be associated with other anomalies. Cases of macroglossia and umbilical hernia associated with NDM are well documented.7-9 Wolcott-Rallison syndrome is an autosomal recessive condition comprising NDM, osteopenia, and epiphyseal dysplasia. Phosphoribosyl-pyrophosphate synthetase hyperactivity, a rare X linked condition, has been described in two brothers with permanent NDM.10 Associations have also been described with coeliac disease,3 intractable diarrhoea, features of hyperimmunoglobulin E syndrome, and absence of the islets of Langerhans,11 and congenital absence of beta cells associated with methylmalonic acidaemia owing to uniparental disomy of chromosome 6.12 A number of disorders of pancreatic organogenesis have been implicated including isolated absence of islet cells and pancreatic hypoplasia or agenesis.13-16 The low IRT in case 2 is suggestive of pancreatic exocrine insufficiency, whereas the abdominal ultrasound and faecal tryptic activity in case 3 are suggestive of the presence of some pancreatic exocrine activity, albeit unquantified. In our three reported cases we could not establish the extent of pancreatic pathology as the parents refused necropsy.
A limited number of disorders of the central nervous system (CNS) have been found in connection with NDM, but not cerebellar agenesis. The CT scan of case 3 and the MRI of case 2 showed cerebellar agenesis. Complete cerebellar agenesis or aplasia is rare.4-6However, familial cases of cerebellar hypoplasia are well documented and show widely variable pathology and modes of inheritance.17 Necropsy would have been helpful to establish the exact nature of the brain malformation in the described cases. CDGS, a condition which may cause cerebellar malformation, was excluded in case 2 as no atypical transferrin glycoforms were detected in the blood.
There have been recent interesting developments in establishing a genetic basis for NDM. In 1995, Temple et al 18 reported an association between transient NDM and paternal UPD6 in two unrelated families and suggested a causal relationship. Furthermore, the finding of an unbalanced duplication of paternal 6q22-23 in a patient with clinical NDM suggested that the critical area of interest lies within this region,2 19and this was supported by a report of an infant with an unbalanced paternal duplication of 6q23-qter who developed NDM.20 It is of note that UPD6 has not been described in association with permanent NDM. UPD6 was excluded in all three cases in this report and there was no evidence of linkage to chromosome 6q24 in this family.
It has been shown that the homeodomain protein insulin promoter factor (IPF1) is critical for development of the pancreas in mice and is a key factor for the regulation of the insulin gene in the β cells of the endocrine pancreas.21 22 Disruption of theIpf1 gene in transgenic mice results in pancreatic agenesis. This has also been shown in man, with a recent report by Stoffers et al 22 of a child with pancreatic agenesis who was homozygous for a single nucleotide deletion in the IPF1 gene, Pro63fsdelC. DNA analysis in our case 1 was negative for this mutation. As pathological examination of the pancreas was not possible in our cases, no definitive conclusions can be drawn as to the extent of the pancreatic abnormality. However, Stoffers et al 22 considered the possibility that the phenotypes of pancreatic hypoplasia and selective agenesis of the endocrine pancreatic islets might represent a spectrum of less severeIPF1 mutations which may impair, but not abolish IPF1 functions. They also suggested that abnormal IPF1 function may be a risk factor for the development of adult onset diabetes mellitus, and in their family, early onset type II diabetes mellitus appeared to cosegregate with heterozygosity for the above mutation.23It is also interesting to note the very strong family history of NIDDM in the cases we have reported. The significance of this is not clear at this stage, but one possible explanation might be that the heterozygotes for the unknown mutation in our family might be at an increased risk of developing diabetes, as were the carriers of theIPF1 mutation described above.
Also of recent interest is the development of the Pax4 knockout mouse. When the Pax4 gene is mutated it causes a lack of pancreatic islet β (and δ) cells, resulting in a lethal autosomal recessive phenotype, characterised by NDM.24Recently it has been proposed that the PAX4gene, located on chromosome 7q32 in humans, is another candidate gene for NDM.25
Diabetes can also be associated with mitochondrial mutations. The A3243G mutation is associated with maternally inherited NIDDM with deafness. Such an association has not been reported in NDM and was excluded in case 2.
The authors are not aware of any reports of associated cerebellar agenesis/hypoplasia and neonatal diabetes mellitus. Findings of both neonatal diabetes mellitus and cerebellar agenesis/hypoplasia in two female sibs and a third female first cousin in a highly consanguineous family is very suggestive of autosomal recessive inheritance.
DNA samples from all three cases and from their immediate family members are being studied with homozygosity mapping, which might indicate the location of the underlying autosomal recessive gene responsible for this condition.
Acknowledgments
The authors are indebted to Doris Stoffers for theIPF1 mutation analysis in case 1, to Dr Rebecca Gardner for the UPD6 analysis, and to Drs Peter Adlard, Anthony Heeley, Fatemeh Hoveyda, Nicholas Mann, and John Wilson for their help, comments, and suggestions.