Article Text

Statistics from Altmetric.com
- ADOA, autosomal dominant optic atrophy
- ADOAC, autosomal dominant optic atrophy and cataract
- AROA, autosomal recessive optic atrophy
- LHON, Leber’s hereditary optic neuropathy
- MGA, 3-methylglutaconic aciduria
- OPA1, optic atrophy 1
- OPA3, optic atrophy 3
- XLAO, X linked optic atrophy
Hereditary optic atrophy is a generic term that refers to a heterogeneous group of genetic disorders for which several modes of inheritance have been described.1 The most common forms of optic atrophy are autosomal dominant optic atrophy (ADOA, OMIM 165500) and Leber’s hereditary optic neuropathy (LHON, OMIM 53500). ADOA, which generally starts in childhood, is characterised by a progressive decrease in visual acuity, blue-yellow dyschromatopsia, loss of sensitivity in the central visual field, and optic nerve pallor.
Mutations in the optic atrophy 1 (OPA1) gene, located on chromosome 3q28–q29, are implicated in about 60–80% of the cases of ADOA.1–4OPA1 encodes for a mitochondrial dynamin related protein. This protein, anchored to the mitochondrial inner membrane, contributes to mitochondrial structure and biogenesis.5,6
A second gene involved in ADOA, not yet identified, has been mapped to chromosome 18q (OPA4, OMIM 605293).7 LHON, which is caused by specific mutations in mitochondrial DNA, is inherited maternally.8 It is characterised by severe bilateral optic atrophy responsible for acute or subacute visual loss, usually starting between the ages of 18 and 35.
Other forms of hereditary optic atrophy include X linked optic atrophy (XLAO, OPA2, OMIM 311050)9 and autosomal recessive optic atrophy (AROA), for which a first locus has recently been mapped to chromosome 8q.10 Finally, more than 15 disorders—mostly inherited in the autosomal recessive mode—have combined optic atrophy and extraocular anomalies. Among these syndromic optic atrophies, type III 3-methylglutaconic aciduria (MGA) (OMIM 258501), also known as the Costeff syndrome11 or the optic atrophy plus syndrome, consists of early onset bilateral optic atrophy, later onset spasticity, extrapyramidal signs, and cognitive deficit. Urinary excretion of 3-methyl glutaconic acid and increased plasma 3-methylglutaric acid levels are the hallmarks of MGA.12 Linkage analyses, undertaken in MGA families originating from inbred Iraqi-Jewish populations, allowed the mapping of the disease causing gene to chromosome 19q13.2–q13.3, close to the gene responsible for myotonic dystrophy.13 The gene responsible for MGA, namely optic atrophy 3 (OPA3), has since been identified.14
Until now, only two mutations in OPA3 have been reported in studies covering nine families affected by type III MGA.14,15 Here we report two missense mutations in OPA3 in two families affected by autosomal dominant optic atrophy and cataract (ADOAC), a disease first described by Garcin et al in 1961 (OMIM 165300).16 Our results indicate that OPA3, encoding a protein in the mitochondrial inner membrane, is responsible for autosomal recessive as well as autosomal dominant optic atrophies.
Key points
-
Hereditary optic atrophies form a heterogeneous group of disorders with different modes of inheritance. OPA1, encoding for a mitochondrial protein, is currently the only gene identified in autosomal dominant optic atrophies. OPA3, implicated in autosomal recessive optic atrophy (OMIM 258501), might also be responsible for autosomal dominant optic atrophy.
-
OPA3 was directly sequenced as a candidate gene in two unrelated families affected with autosomal dominant optic atrophy and cataract (ADOAC, OMIM 165300). OPA3 was studied in 11 affected individuals and 10 healthy relatives. In addition, OPA3 encodes for a mitochondrial protein of unknown function, possible mitochondrial dysfunction in skin fibroblasts from a patient with ADOAC was investigated.
-
Two different mutations in exon 2 of OPA3 were found in patients with ADOAC. The 277G→A (G93S) mutation was present in one family, and the 313C→G (Q105E) mutation in the other. Both mutations segregated with the disease in each family and were absent in healthy relatives and in 400 control chromosomes. No abnormalities were found in the respiratory chain or in the mitochondrial membrane potential, or in the organisation of the mitochondrial network of the fibroblasts obtained from one affected patient. However, the fibroblasts showed increased susceptibility to staurosporine induced apoptosis.
-
The results indicate that OPA3 is responsible for dominant optic atrophy as well as autosomal recessive atrophy. Thus, along with autosomal dominant optic atrophy and Leber’s hereditary optic neuropathy, ADOAC is yet another form of hereditary optic atrophy involving a mitochondrial inner membrane protein and a possible apoptotic mechanism.
METHODS
Clinical report
Family 1
We re-examined the French family first reported in 1961 by Garcin et al,16 in which members belonging to four generations were affected by ADOAC (fig 1).

Pedigrees of the families affected by autosomal dominant optic atrophy and cataract (ADOAC). Filled symbols = affected; empty symbols = unaffected; square = male; circle = female; star = subjects in whom the OPA3 gene was sequenced.
Patient III.3, a female born in 1920, suffered from a visual impairment from infancy which worsened from the age of 28. At age 38, her visual acuity was 2/10 in both eyes and ophthalmological examination revealed bilateral optic atrophy and posterior cortical cataract. On neurological examination there was resting and postural tremor of both hands and a mild extrapyramidal rigidity of both upper limbs. She had bilateral pes cavus and absence of deep tendon reflexes in the lower limbs. The cataract was operated on at age 51, when her visual acuity was 1/10 in each eye. Neurological examination at age 83 showed the same extrapyramidal signs as before, but there was no worsening of the disease.
Patient IV.1, a female born in 1943, suffered from decreased visual acuity before the age of 10. At age 15, she had a visual acuity of 5/10 in each eye, bilateral optic atrophy, and bilateral posterior cortical cataract. Neurological examination at age 15 showed mild postural tremor, mild rigidity of both upper limbs, and mild bilateral pes cavus. Her visual acuity gradually decreased to 1/10 in both eyes. Cataract lens implant surgery was done at the ages of 47 and 48. At age 58, she developed bilateral central scotomata. Neurological examination at age 60 showed a mild postural tremor of the upper extremities.
Patient IV.2, a female born in 1945, contracted paralytic poliomyelitis at age 18 months. Her visual acuity first decreased before the age of 10. At age 13, the visual acuity was 3/10 and 9/10 in the right and left eyes, respectively. Fundus examination revealed bilateral optic atrophy, and slit lamp examination showed bilateral anterior cortical cataracts. Cataract lens implant surgery was done at the ages of 45 and 46. Visual acuity was 2.5/10 and 3.2/10 in the right and left eyes, respectively, at age 57. At that age, neurological examination, although difficult because of sequelae of poliomyelitis, showed postural tremor without additional extrapyramidal signs. Magnetic resonance imaging of the brain was normal.
Patient V.1, a female born in 1967, first had a decrease in visual acuity at the age of 12. At age 18, visual acuity was 4/10 in each eye. There was bilateral optic atrophy on fundus examination, and slit lamp examination showed bilateral anterior cortical cataracts. Bilateral central scotomata were demonstrated by Goldmann visual field examination. The Farnsworth-Munsell colour vision test revealed a mild red-green dyschromatopsia. Electroretinography was normal and visual evoked potential (VEP) testing showed a bilateral increase in P2 latency (120 to 158 ms). Cataract lens implant surgery was undertaken at age 25. At age 29, the visual acuity was 2/10 in the right eye and 3/10 in the left eye. Neurological examination was normal.
Patient VI.1, a female born in 1995, was first examined at age three years and eight months for visual impairment. Fundus examination was normal but the slit lamp showed bilateral anterior and posterior subcapsular opacities. Cataract surgery was done at four years of age. Three months after lens surgery, the visual acuity was 7/10 in each eye. Though the fundus appeared pale, it was still considered normal. VEP at age 7 were normal in both eyes. The neurological examination was normal.
Patient IV.6, a female born in 1953, had visual impairment from early infancy. The diagnosis of optic atrophy was made in infancy and she received special education for visually impaired children. She currently has lens opacities but cataract surgery has never been done. She has no neurological signs at age 50.
Patient V.6, a female born in 1982, has also had visual impairment from early infancy but went through normal schooling. Cataract surgery was done at age 5. Neurological examination was normal at age 5.
Detailed clinical data are unavailable from the other patients in the same family affected with ADOAC—that is, patients IV.5, V.3, V.4, and VI.3.
Family 2
Patient III.3, a female born in 1944, suffered a decrease in visual acuity from age 12. At age 37, her visual acuity was 1/50 with a central scotoma and optic nerve atrophy in each eye. She became legally blind at age 54, and posterior capsular cataract was diagnosed at age 56.
Patient III.7, a female, was born in 1955. Her visual acuity decreased from age 10. Bilateral cataract was diagnosed at age 45, and she is currently legally blind.
Patient IV.1, a female born in 1971, suffered a decrease in visual acuity from age 6. At age 9, her visual acuity was 4/10 with a central scotoma and blue-yellow dyschromatopsia in both eyes. Electroretinography was normal. A bilateral cataract was diagnosed at age 10.
Ophthalmological examination at age 33 showed a visual acuity of 0.5/10 in both eyes. Fundus examination revealed bilateral atrophy and temporal pallor of the optic nerve. Slit lamp examination showed bilateral cerulean cataracts (fig 2). Neurological examination was normal.

Cerulean cataract observed in patient IV.1 from family 2.
Patient IV.2, a male born in 1976, was affected by optic atrophy at age 12. At that age, his visual acuity was 3/10 in each eye, and he had bilateral central scotomata and blue-yellow dyschromatopsia. Surgery for bilateral capsular posterior cataract was done when he was 19.
OPA3 gene sequencing
Blood samples were obtained from eight affected patients and eight healthy relatives from family 1 and three affected patients and two healthy relatives from family 2.
Genomic DNA was extracted from blood samples using the High Pure polymerase chain reaction (PCR) template preparation kit (Roche Diagnosis, Mannheim, Germany). Four primer couples were used to amplify the two coding exons.14 PCR reactions were carried out under standard conditions with 100 ng of genomic DNA in a 50 µl volume: 1.5 mM MgCl2, 75 mM Tris-HCl (pH 9 at 25°C), 20 mM (NH4)2SO4, 0.01% Tween 20, 50 pmol of each primer, 200 µM of each dNTP, and 2 units of Hot GoldStar (Eurogentec, Seraing, Belgium). Each of the 30 cycles consisted of a denaturation step of 30 seconds at 94°C, a hybridisation step of 30 seconds at 58°C, and an extension step of one minute at 72°C. The purified PCR products were sequenced using a Ceq2000 DNA sequencer (CEQ DTCS-Quick Start Kit, Beckman Coulter, Fullerton, California, USA). Multiple sequence alignments were made using CLUSTALW software.
LOD score calculations
Multipoint LOD score analysis was undertaken in family 1 using markers D3S1601, D3S3669, and D3S2748 at the OPA1 locus with the Genehunter computer package under an autosomal dominant model of transmission, with a maximum disease frequency of 1/12 000,2 as well as in a non-parametric model.
Biochemical investigations
The plasma concentrations of lactate, pyruvate, and ketone bodies were assessed by spectrophotometry using commercial kits (Roche Diagnostics, Meylan, France). Urinary organic acids and plasma amino acids were analysed, respectively, by gas and ion exchange chromatography. The activity of the fibroblast mitochondrial respiratory chain was assessed by the polarographic measurement of oxygen consumption and by the determination of the individual activity of the respiratory chain complexes II, III, and IV, according to Rustin et al.17
Apoptosis
Fibroblasts, obtained from patient IV.2 by skin biopsy, were cultured in RPMI 1640, 10% fetal calf serum, and 5% CO2. The fibroblasts were exposed to staurosporin at concentrations of 50 or 100 nM over 24 hours. The percentage of apoptosis was calculated from triplicate values obtained in the course of two independent experiments; apoptotic nuclei were identified by the presence of condensed chromatin on DAPI staining. Control fibroblasts were obtained from an unaffected individual and prepared at the same time and in the same way as the patient’s cell line.
Mitochondrial membrane potential
For the analysis of the mitochondrial membrane potential (ΔΨm), the fibroblasts were incubated for 20 minutes with 5 µg/ml JC-1 (Molecular Probes, Leiden, Netherlands) in the culture medium, then collected, washed in phosphate buffered saline (PBS), and processed for fluorescence analysis on an Ascent Fluoroscan using the Ascent Software (excitation 488 nm; emission 538 nm and 590 nm). The ratio of the fluorescence at 590 nm, proportional to the ΔΨm, to that at 538 nm, proportional to the mitochondrial mass, was calculated from triplicate values obtained in the course of two independent experiments.
Mitochondrial network
Cells grown on glass coverslips were stained directly in the culture using 100 nM CMXros Mitotracker® Red (Molecular Probes) for 30 minutes, then fixed and DAPI stained in PBS, 3.7% paraformaldehyde (30 minutes, 4°C). Fluorescence images were captured and processed using a Leica DMIRE-2 microscope.
RESULTS
Families 1 and 2 relate to a group of 25 families affected by dominant optic atrophy for which negative results in sequencing OPA1 had previously been obtained in our laboratory. Furthermore, the OPA1 locus was excluded by linkage analysis in family 1 on the basis of a significantly negative LOD score of −4.83 at θ = 0. The OPA3 gene was subsequently studied in both families with reference to a candidate gene.
We found two novel missense mutations in exon 2 of the OPA3 gene—namely, the 277G→A (G93S) mutation in all affected members from family 1, and the 313C→G (Q105E) mutation in those of family 2 (fig 3). In both families, the mutations segregated with the disease and were absent in healthy relatives as well as in 400 control chromosomes. Both these mutations of the OPA3 gene involve strongly conserved residues in the OPA3 orthologs of different species (fig 4).

Electropherograms of OPA3 gDNA sequencing showing wild-type (WT) and mutant (MT) sequences in, respectively, healthy (IV-3) and affected members of family 1 (IV-2 and V-1) and healthy (IV-3) and affected member (IV-1) of family 2. Direct and reverse sequences are given for patient IV-1 of family 2.

Amino acid alignments of different orthologs of OPA3 genes (Clustal W 1.82). The box shows the amino acid conserved in all five species.
No abnormalities of the respiratory chain (data not shown), the mitochondrial membrane potential (fig 5), or the morphology of the mitochondrial network (fig 6) were found in fibroblasts from patient IV.2 from family 1. Lactataemia and urinary concentrations of 3-methylglutaconic and 3-methylglutaric acids were not increased in patient IV.2 (data not shown). Nevertheless, an increased susceptibility to the induction of apoptosis was observed in cultured fibroblasts treated with staurosporin. Indeed, an average of 35% of apoptotic cells was found in the fibroblasts of the affected individual, versus 5% in controls (fig 7).
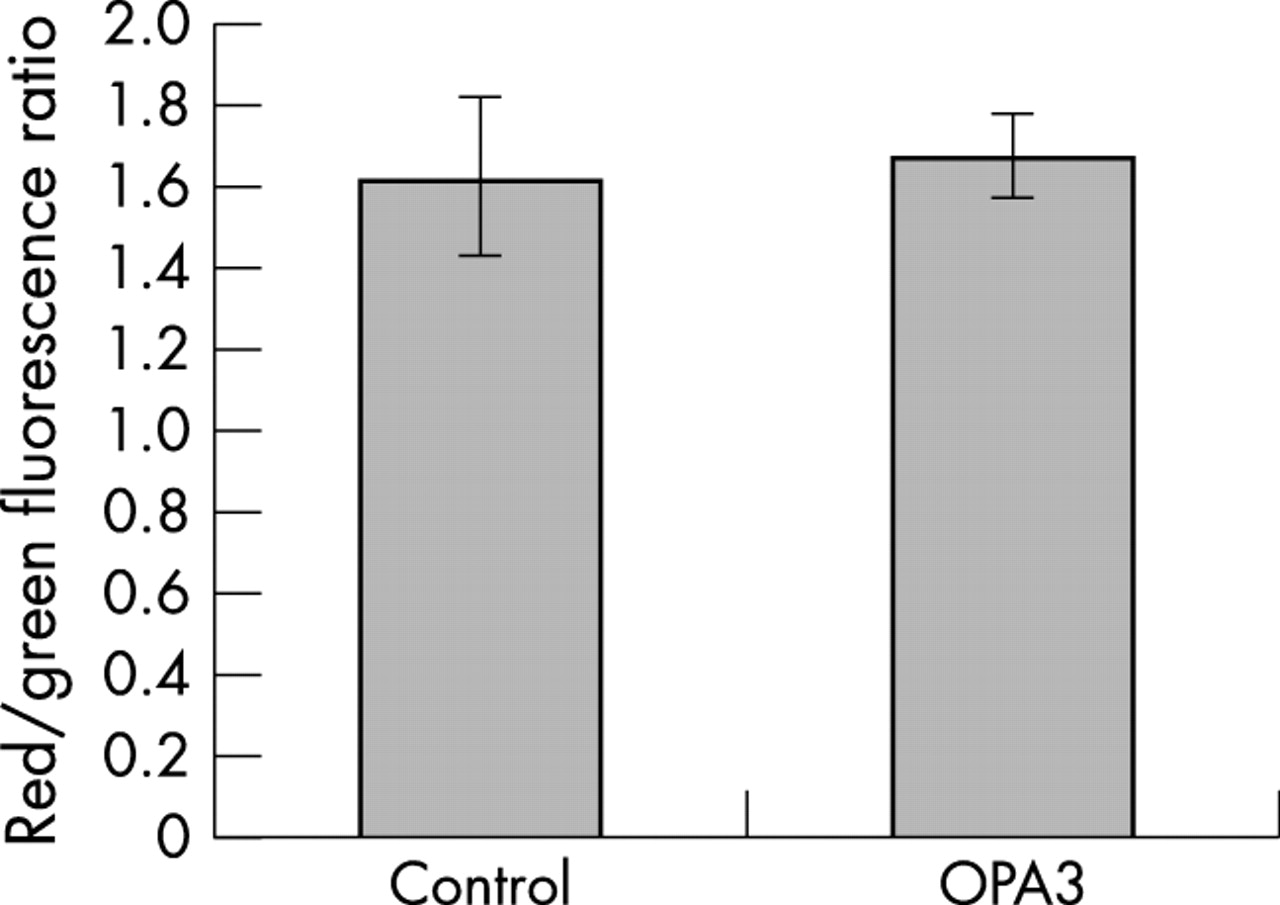
ΔΨm assessment with JC-1 dye in fibroblasts from patient IV-2 (family 1) versus control fibroblasts.

Mitochondrial network morphology. Fibroblasts from patient IV-2 (right); control fibroblasts (left).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Staurosporin induced apoptosis in fibroblasts from patient IV-2 versus control fibroblasts. Average values calculated from triplicate values obtained in the course of two independent experiments. Light grey, 50 mM staurosporin; dark grey, 100 mM staurosporin.
DISCUSSION
We report here the molecular basis of an autosomal dominant disorder referred to as optic atrophy, cataract, and neurological disorder in the OMIM database (165300), first described by Garcin et al in 1961.16 Although these investigators described the disease as a neurodegenerative disorder and discussed a possible relation to Friedreich’s ataxia, the neurological signs are mild or even absent in affected patients. Thus the disease could be more appropriately designated as autosomal dominant optic atrophy and cataract (ADOAC).
ADOAC is caused by a mutation in the OPA3 gene, known to be involved in type III 3-MGA, a neuro-ophthalmological syndrome consisting of early onset bilateral optic atrophy, later onset spasticity, extrapyramidal dysfunction, cognitive deficit, and abnormal urinary excretion of 3-methylglutaconic acid and 3-methylglutaric. The OPA3 gene contains two exons and its main transcript (∼5 kb) is ubiquitously expressed. It encodes a peptide composed of 179 amino acids displaying a putative mitochondrial N-targeting signal sequence and a C-terminal coiled-coil domain.14 A proteomic study, done on mouse liver, has recently shown that the OPA3 protein is located in the inner mitochondrial membrane,18 but its function remains unknown. Only two mutations in the OPA3 gene have been reported so far (table 1). One of these—a homozygous splice site mutation (IVS1-1G>C) abolishing mRNA expression in fibroblasts—was first found in Iraqi-Jewish patients with type III MGA.14 The other mutation, corresponding to a homozygous deletion in exon 2 (320–337del), resulting in deletion of six amino acids between residues 108 and 113, was reported in a Kurdish-Turkish patient diagnosed with type IV MGA (3-methylglutaconicaciduria, severe psychomotor retardation, and cerebellar dysgenesis).15
Mutations reported in the OPA3 gene
The novel two mutations of the OPA3 gene reported here were both heterozygous substitutions in exon 2. Several arguments support the pathogenicity of these mutations. First, both mutations segregated with the disease in each pedigree through different generations, and both were absent in a set of 400 control chromosomes. Second, both involved two strongly conserved residues in the OPA3 orthologs. Third, the mutation 277G→A increased the susceptibility to apoptosis induced by staurosporine in cultured fibroblasts. Thus different mutations in the OPA3 gene may be responsible for autosomal recessive or autosomal dominant optic atrophies. Homozygous mutations in OPA3 with loss of function lead to a severe multisystem disease with optic atrophy, whereas a heterozygous missense mutation results in a milder phenotype.
Interestingly, heterozygotic parents of patients affected by type III MGA are not affected by ADOAC, suggesting that haploinsufficiency of the OPA3 gene probably has milder consequences. Conversely, the effects of the missense mutations reported here are responsible for ADOAC in heterozygous patients. This would suggest a dominant negative effect of the G93S and Q105E OPA3 mutant alleles.
OPA1 and OPA3 genes, responsible for ADOA and ADOAC respectively, are currently the only nuclear genes identified in dominantly inherited optic atrophy. Interestingly, three forms of hereditary optic atrophy for which the molecular bases are known involve either the mitochondrial DNA (LHON) or nuclear genes coding for mitochondrial proteins (OPA1 and OPA3). Comparison between LHON, ADOA, and ADOAC also reveals similarities at the clinical and molecular levels, as follows.
First, in all three diseases, optic atrophy can be associated with more complex neurological signs such as dystonia or the Leigh-like syndrome in LHON,19,20 deafness in ADOA,21 and mild extrapyramidal signs in ADOAC.
Second, all three diseases are caused by mutations in genes encoding proteins located in the inner mitochondrial membrane. LHON is mainly caused by mutation of mitochondrial DNA genes encoding respiratory chain complex I subunits; the OPA1 gene encodes a protein anchored to the mitochondrial inner membrane facing the intermembrane space6; and the OPA3 protein is also located in the mitochondrial inner membrane.18
Third, LHON, ADOA and ADOAC are all associated with painless optic atrophy without inflammation, consistent with an apoptotic mechanism, which could be common to the three disorders causing retinal ganglion cell death culminating in optic atrophy. Indeed, it has been shown that complex I plays a key role in the permeability of transition pores22 and that cells bearing mutations causing LHON are predisposed to apoptosis,23,24 possibly because of increased production of reactive oxygen species.25 Similarly, it has been shown that the silencing of OPA1 by RNA interference in HeLa cells leads to cytochrome c release and commits cells to apoptosis.26
Finally, increased susceptibility to the induction of apoptosis was also observed in our study in fibroblasts carrying the G93S mutation in OPA3. The further presence of cataract in ADAOC could also be explained by an apoptotic mechanism since there are several lines of evidence implicating apoptosis in cataractogenesis.27
Conclusions
The OPA3 gene coding for a protein of unknown function of the mitochondrial inner membrane is responsible for both autosomal recessive and dominant optic atrophy. Further studies will be required to determine the role of the OPA3 protein in promoting increased cell death and to define the involvement of the OPA3 gene in other forms of optic atrophy.
Acknowledgments
We sincerely thank all the patients who have helped us in our research. We are grateful to Dr Jean-Paul Loemba for ophthalmological examination of patients from family 2, Yves Tourmen for bioinformatic searches, Philippe Bonnaud, Anne Coutolleau, Dominique Couturier, and Florence Fatih for excellent technical assistance, and Kanaya Malkani for critical reading and comments on the manuscript. The work was supported by INSERM, by GIS-Institut des Maladies Rares, the University Hospital of Angers (CHU), the University of Angers, France, by Retina France, and the Association Française contre les Myopathies. CB was awarded a grant by the “Fondation pour la Recherche Médicale”.
REFERENCES
Footnotes
-
↵* The first two authors contributed equally to this work
-
Conflicts of interest: none declared