Article Text

Statistics from Altmetric.com
Key points
-
The objective of this study was to review, using Medline, all cases with supernumerary marker chromosomes (SMC) and uniparental disomy (UPD), and to reflect on the aetiology and mechanisms of formation of SMC.
-
The survey showed only 19 cases, a maternal UPD(1), a paternal UPD(6), a maternal UPD(7), a maternal UPD(9), a maternal UPD(10), a maternal UPD(12), a maternal UPD(20), and a maternal UPD(22), as well as eight cases with maternal UPD(15) and three cases with paternal UPD(15).
-
Mechanisms of formation assumed are functional rescue by mitotic reduction of the monosomic homologue to a SMC in a trisomic zygote; somatic reduplication of the normal homologue in a 46,XN,mar zygote; completely mitotic formation by non-disjunction and subsequent reduction of the monosomic homologue or vice versa; and fertilisation of a disomic gamete by a gamete already carrying a marker chromosome. Apart from the first, all these mechanisms increase the likelihood for UPD being present in cases with a SMC.
-
Mechanisms of formation seem to be different in additional inv dup(15), other additional isochromosomes, and SMCs characterised by one or two deletions. The low incidence of SMC associated with UPD, the formation of additional inv dup(15), and other isochromosomes, as well as the results of molecular investigations of trisomy 21 mosaicism are all evidence against an active mechanism creating a SMC in a trisomic zygote.
Uniparental disomy (UPD) describes the inheritance of a pair of chromosomes from only one parent.1 Mechanisms of formation are trisomy rescue, monosomy rescue, post-fertilisation errors, and gamete complementation. Problems associated with UPD include trisomy mosaicism, genomic imprinting, homozygosity of autosomal recessive mutations, or even a combination of these.2 Supernumerary marker chromosomes (SMCs) are predominant in only a proportion of cells leading to a specific kind of trisomy mosaicism. The presence of a cell line with two normal chromosomes and a second hyperploid cell line promoted speculation that people with a SMC might have an increased risk for UPD of the structurally normal homologues from which the SMC was derived.3
In the present article, published reports of UPD associated with a SMC as well as fundamental aspects of chromosome segregation and UPD are reviewed. Additionally, two questions will be discussed. Is the association of UPD with a SMC only coincidence or the consequence of an active mechanism? Which mechanisms are most likely for the formation of a SMC?
METHODS AND RESULTS
An extensive search in the Medline database showed only 19 cases of a SMC associated with UPD. Maternal UPD was found in 15 cases and paternal UPD in four cases (table 1). One case each was found for chromosomes 1, 6, 7, 9, 10, 12, 20, and 22,4–11 and 11 cases for chromosome 15, eight of them with Prader-Willi syndrome and three of them with Angelman syndrome, were found.3,12–19 FISH investigations of one of the maternal UPD(15) cases indicated X chromosomal origin of the SMC.14 In another case, maternal UPD(22) was segmental and associated with a de novo 11;22 translocation.10 Analysis of the molecular results showed only two isodisomic cases in 10/15 of the maternal UPD group6 and also only one heterodisomic case in 3/4 of the paternal UPD group.18 No molecular results delineating heterodisomy versus isodisomy were reported in the remaining cases.
Single case reports of autosomal uniparental disomy associated with a supernumerary marker chromosome
A survey on systematic searches for UPD associated with a SMC other than inv dup(15) in postnatally ascertained cases showed only retrospective and therefore biased studies with two out of 49 cases testing positively,5,7,20–23 one case with paternal UPD(6),5 and one case with maternal UPD(9) (table 2).7 In eight postnatal searches for UPD(15) in a total of 122 cases with an additional inv dup(15) and various phenotypes,13,24–30 only two positive cases were found (table 3).13,26 In one prenatal study, only two cases with UPD(15) out of 26 cases with an additional inv dup(15) were found (table 3).15
Systematic searches for uniparental disomy in cases with a supernumerary marker chromosome (positive cases in brackets)
Systematic searches for uniparental disomy in cases with an additional inv dup(15) (positive cases in brackets)
Prenatal incidence of SMC is approximately 1:2500 and approximately 30-50% originate from chromosome 15.31,32 The incidence of specific markers other than inv dup(15) is not known. Most studies were performed before FISH with centromeric probes was established, and therefore the number of SMCs of unknown chromosomal origin is extremely high.32
DISCUSSION
Coexistence of UPD with a SMC has mainly been explained by the following four mechanisms (fig 1).3
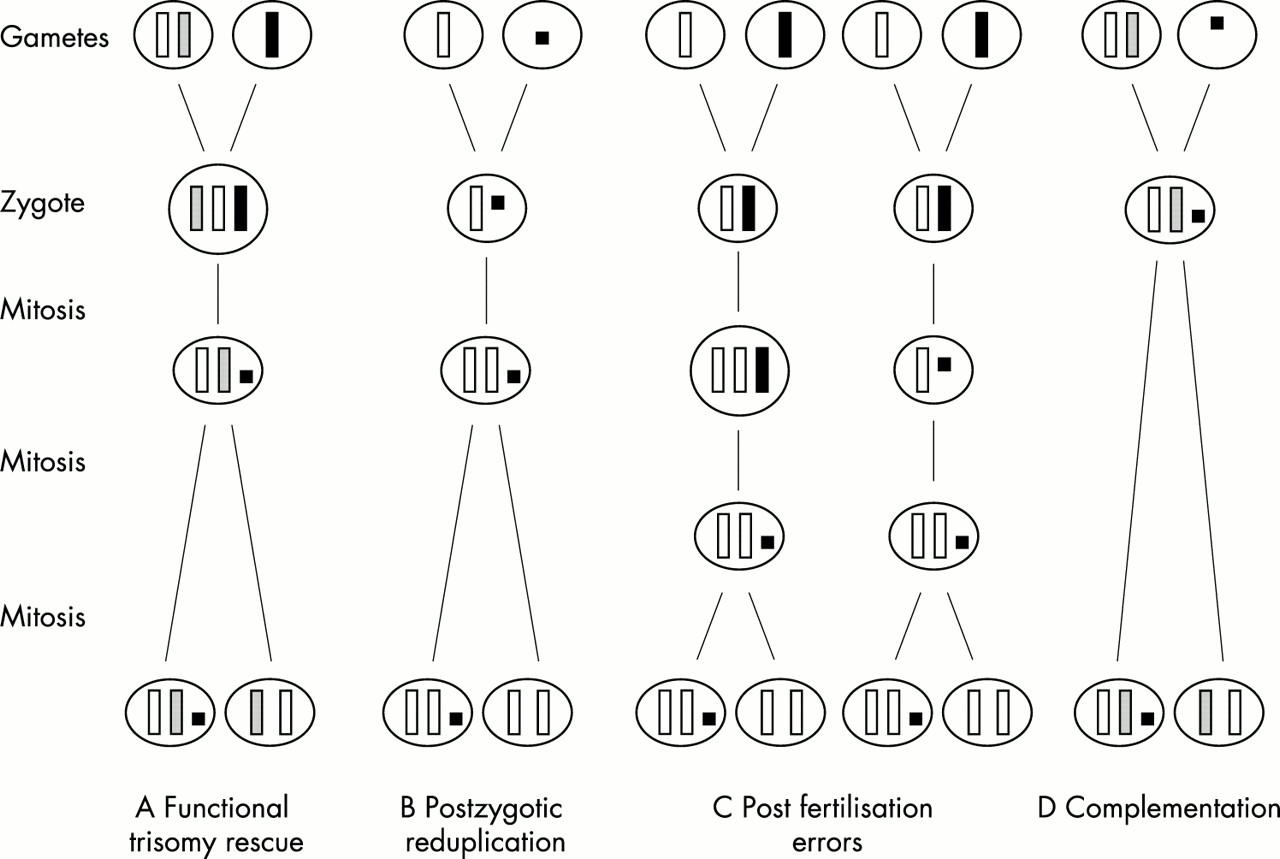
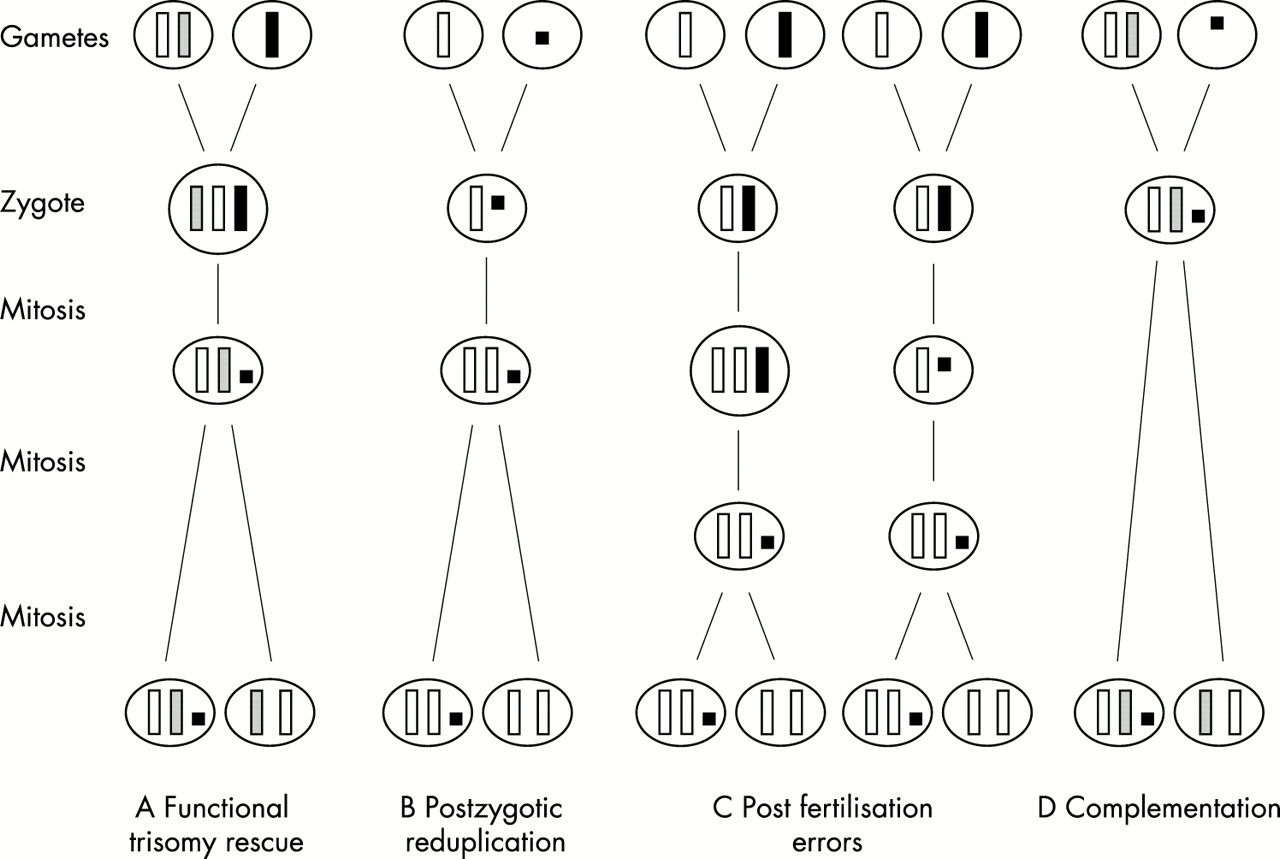{kind=link}
Mechanisms of formation of UPD associated with a SMC. (A) Functional trisomy rescue by mitotic reduction of the single homologue in a trisomic zygote. (B) Postzygotic reduplication by fertilisation of a normal gamete by a gamete carrying a SMC followed by somatic reduplication of the normal homologue. (C) Post-fertilisation errors by either mitotic non-disjunction followed by reduction of the single homologue or vice versa. (D) Complementation by fertilisation of a disomic gamete by a gamete carrying a SMC.
-
The zygote may have originated as a trisomy with parts of the single parental homologue having been lost through one or two breakage events in an early embryonic cell division (“functional trisomy rescue”), either at random or even by an active mechanism (fig 1A). Here, two subclasses can be delineated. The first is characterised by two breakage events in the proximity of the centromere. In the case of occurrence in a very early cell division and of a UPD not affecting the phenotype, clinical outcome should be normal, because only heterochromatic material should be present. Such cases will be detected only by chance or by strictly prospective studies. The second subclass is characterised either by one breakage resulting in, strictly speaking, a deleted chromosome and not a classical marker chromosome or by two breakages more or less distal to the centromere. Owing to the euchromatic genetic material involved, both will create a clinically relevant phenotype, irrespective of the presence or absence of UPD. Survival should correlate inversely with the size and the chromosomal origin of the marker.
-
Duplication of the normal homologue in a zygote which has inherited a SMC in place of the normal corresponding chromosome (karyotype 46,XN,mar) “rescuing” an aneuploidy (fig 1B). In this case, UPD arises by “postzygotic reduplication” and therefore complete isodisomy should always be observed. This mechanism seems not to be frequent. Only four cases with isodisomy out of 19 cases investigated were reported.3,5,8,18 Three of them were of paternal origin,3,8,18 reflecting the general rule that isodisomy is more likely in paternal than in maternal UPD.2 This low number argues against an active mechanism.
-
A completely postzygotic formation by either non-disjunction in an early mitosis and subsequent reduction of the monosomic homologue or by an inverse sequence (“post-fertilisation error”) (fig 1C). This subclass is the most complex and hard to demonstrate, and no cases have been reported so far.
-
Fertilisation of a disomic gamete by a gamete with a SMC formed before or during meiosis (“complementation”) (fig 1D). Combined incidence of SMC and UPD is very low, but both figures are strongly biased by selection owing to survival and by the method of ascertainment. However, illustrated by the presence of large polymorphic satellites on the short arm of one chromosome 15 of the father and on the inv dup(15) of the affected daughter, this mechanism is most likely in a case with maternal UPD(15) and an additional inv dup(15).17 The same mechanism can be assumed in a case of maternal UPD(15) and an additional marker of X chromosomal origin.14 Otherwise, a double aneuploidy simultaneous with subsequent reduction of one X chromosome and loss or lack of the paternal chromosome 15 must be postulated.
In nine out of the 12 cases investigated by molecular methods, only the mechanisms of “functional trisomy rescue” and “complementation” could explain the outcome. In neither mechanism can it be differentiated whether the SMC was formed before or during meiosis or in an early somatic cell division. If it was formed before or during meiosis, the coexistence of a SMC with UPD would be only coincidental. Two independent events, non-disjunction in the meiosis of one parent and aberrant segregation or even formation of the SMC in the meiosis of the other parent, must have occurred. The question of which mechanism is more likely cannot be answered definitely. However, the mechanism of “functional trisomy rescue” is the more complex one and there are some arguments against it, particularly against an active mechanism. (1) Mosaicism in the placenta or even in the fetus with a cell line trisomic for a whole chromosome and not detected by routine investigations but relevant to the clinical outcome is possible. No cases with similar markers but discrepant clinical outcome have been reported so far. (2) Trisomy 21 has been observed in approximately one out of 700 newborns,34 but additional der(21) was not reported more frequently than other SMCs. In trisomy 21 mosaicism subsequent to a trisomic zygote, there is no preference to lose the single homologue. Exactly one third of all cases investigated lost the single homologue, whereas in the remaining cases one of the two uniparental homologues was removed.34 The latter argues against an active mechanism focused on the monosomic homologue. (3) Trisomy 9, 12, 15, 18, and 22 more often result in an isochromosome rather than in a more simple reduction to a SMC. So far, no cases of UPD have been reported in cases with one of the common additional autosomal isochromosomes, inv dup(22q) and i(9p), i(12p), or i(18p). Most of the latter are caused by maternal meiosis II non-disjunction and a subsequent mitotic formation of the isochromosome.35,36 UPD(18) has not been reported at all. Maternal UPD(9) and maternal as well as paternal UPD 22 are associated with a normal outcome,37–39 but so far no cases of UPD associated with an i(9p) or an inv dup(22q) have been reported. (4) In a prenatal and therefore not phenotypically biased study investigating cases with an additional der(15), only two out of the 26 cases tested positive for UPD(15).15 Therefore, assuming a trisomic zygote at least in these cases there should be no preference for SMC formation of the monosomic homologue. Moreover, why is not the whole monosomic homologue eliminated assuming the presence of an active rescue mechanism?
In the case of an inv dup(15), the situation seems to be the reverse. Only seven cases with Prader-Willi syndrome and three cases with Angelman syndrome, all associated with UPD(15) shown by molecular methods, have been reported.3,12–13,15–19 This figure is unexpectedly low for several reasons. The frequency of cases with an additional inv dup(15) is high and in contrast to the frequency of UPD in case reports as well as in systematic studies of UPD(15) associated with an inv dup(15) (two positive out of 122 postnatal cases and two out of 26 prenatal cases) (table 3). This is particularly low considering the general ascertainment bias of reporting positive cases of an inv dup(15) associated with UPD(15). Therefore, in trisomic zygotes transforming one homologue into an inv dup(15), there is no preference for the monosomic homologue. Most likely different mechanisms are responsible for an inv dup(15) associated with biparental disomy 15 and for an inv dup(15) associated with UPD(15). In the case of UPD(15) the inv dup(15) might be formed before or during meiosis and both the UPD and the marker are already present in the zygote.16 In contrast, in cases with biparental disomy 15 associated with an inv dup(15), the zygote was trisomic and the marker more likely arose mitotically preventing UPD(15).
It is obvious that the reports of SMC associated with UPD are extremely biased. Almost all investigations have been carried out as a consequence of an abnormal phenotype. No large or systematic prenatal studies have been reported so far. However, the results of such studies and, in addition, a follow up of the cases detected there would greatly assist genetic counselling.
In conclusion, the data on UPD associated with a SMC are scarce. At this time, there are no hints towards an active mechanism creating a SMC in a trisomic zygote, but the incidence of UPD in cases with a SMC is increased by coincidence, ascertainment bias, and because for most chromosomes only then is the fetus viable. Further reports of cases with a SMC both associated and not associated with UPD and particularly more systematic studies would provide more information on a topic extremely relevant for prenatal genetic counselling. Therefore, after careful genetic counselling, molecular investigations for UPD should be performed more often in cases with a SMC, particularly in chromosomes for which genomic imprinting is known or at least assumed.