Article Text

Statistics from Altmetric.com
- LWD, Leri-Weill dyschonrdosteosis
- BMD, bilateral Madelung deformity
- SDS, standard deviation scores
- BMF, bone marrow fibroblasts
- SNP, single nucleotide polymorphism
- PAR, pseudoautosomal region
In 1878, Madelung described a painful, disabling, and deforming abnormality of the forearm in which misalignment of the bones of the wrist and forearm resulted in a “spontaneous forward subluxation of the wrist”.1 A familial form of a bilateral wrist deformity resulting from dorsal dislocation of the ulnar head, accompanied by mesomelic short stature was first reported in 1929 and later termed Leri-Weill dyschondrosteosis (LWD).2–4 In LWD, the Madelung deformity results from dysplasia located at the medial aspect of the distal radial growth plate, where narrowing and premature fusion of the physis occurs.5 This dysplasia leads to differential growth rates between the lesion and the distal lateral radial physis impeding growth and pulling the lateral radial epiphysis off line as the remaining physis advances.6 In its fully developed form, Madelung deformity leads to shortening and bowing (sometimes marked) of the radius and the involvement of other epiphyses in this disorder results in mesomelia.7 Typically, Madelung deformity does not appear until mid-childhood but is usually preceded by radiological signs.8 Other localised lesions in the ulnar-palmar zone of the distal radius can result in differential growth rates leading to Madelung deformity including Turner, Olliers, and multiple exostoses syndromes and environmental causes.3,4,9
Key points
-
Haploinsufficiency of the short stature homeobox containing gene (SHOX), which maps to the pseudoautosomal region (PAR1) of the X and Y chromosomes, is the molecular basis of Leri-Weill dyschondrosteosis (LWD). We undertook the mutation and expression analysis of this gene in a cohort of 18 probands with bilateral Madelung deformity (BMD).
-
Madelung deformity occurs as the result of dysplasia of the distal radial growth plate and is a distinctive clinical feature of LWD. In our cohort, comprising both familial and sporadic BMD probands, mutations in SHOX were identified in 12/18 probands. These included six large scale deletions detectable by FISH, three point mutations (R195X, Y35X, and IVS2-1G>A), of which the latter two are novel, and three possible 5‘ deletions/rearrangements identified by segregation analysis of null alleles, within and flanking SHOX.
-
Analysis of SHOX expression by RT-PCR in patient bone marrow fibroblasts confirmed that monoallelic SHOX expression was present in two probands. The first had the IVS2-1G>A point mutation and had inherited the null allele at the PAR1 SHOX flanking marker, DXYS233. Two further families with dominant transmission of Madelung deformity cosegregated for LWD and null alleles at SHOX flanking/intragenic markers.
Although LWD appeared to be an autosomal dominant disorder, patients with X/Y chromosomal rearrangements suggested a X/Y homologous locus for the gene.3,4 Subsequently, the isolation of the short stature homeobox containing gene (SHOX) from a distal Xp short stature gene interval10,11 was found to be coincident with the Xp22.3 assignment for the LWD locus.12,13 Thereafter identification of SHOX mutations in LWD established the molecular basis of this disorder.12,13 In this communication, we report an investigation of the molecular pathogenesis of Madelung deformity of childhood by undertaking SHOX mutation analysis and tissue specific expression of this gene in a cohort of 18 probands manifesting bilateral Madelung deformity (BMD).
MATERIALS AND METHODS
Patients
Following ethical approval, probands were recruited before or after undergoing corrective physiolysis/Vickers procedure for Madelung deformity between 1979 and 2000, at the Royal Children’s Hospital, Brisbane. Measurements of standing heights were converted to standard deviation scores (SDS).14,15 Following informed consent, blood samples were taken from probands and available family members for metaphase preparation and DNA extraction. When available at physiolysis, bone marrow fibroblasts (BMFs) were harvested.
Karyotype and fluorescence in situ hybridisation (FISH) analyses
Karyotypes were determined by standard methods.16 Fluorescence in situ hybridisation (FISH) analysis was carried out in metaphases, using the cosmid LLNOYCO3‘M‘34F5, which contains exons III to VIb of SHOX.10,17 This cosmid was biotin labelled using the Bio-Nick labelling system (Invitrogen, Melbourne) and hybridised to metaphase spreads of patient lymphocytes using standard protocols. After post-hybridisation washes, the slides were incubated with mouse monoclonal anti-biotin antibody (DAKO, Carpinteria, CA) and then detected with Oregon Green labelled goat anti-mouse polyclonal antibody (Molecular Probes, Eugene, OR). Cohybridisation was undertaken using the X centromeric (DXZ1) and Y centromeric (DYZ3) probes (Vysis Inc, Downer’s Grove, IL).
PCR amplification and SHOX sequencing
Genomic DNA was isolated from peripheral lymphocytes by standard methods.18 Except where indicated, all PCRs were performed in 50 μl volumes, containing 1 μg of genomic DNA, 0.4 μmol/l of each primer, 20 mmol/l Tris/HCl, pH 8.4, 50 mmol/l KCl, 0.2 mmol/l each dNTP, 1.5 mmol/l MgCl2, and 2.5 units of Taq polymerase (Invitrogen, Melbourne). PCR products were purified using the QIAquick PCR purification kit (Qiagen, Aust) and sequenced using the BigDye terminator cycle sequencing ready reaction kit (Applied Biosystems, Melbourne). SHOX exons II to VIa, containing the entire coding sequence, were amplified separately with 30 cycles of PCR using previously described primer sequences and conditions,10 except as follows. Exon II was amplified in a single reaction to yield a 430 bp product substituting AmpliTaq Gold (Roche, Sydney) for Taq polymerase (Invitrogen, Melbourne), and cycling conditions were 50 cycles of 94°C/58°C/72°C for 30 seconds each. Exons IV and V were amplified together as a single 334 bp fragment at an annealing temperature of 58°C using the exon IV sense and exon V antisense primers.10 Dimethyl sulphoxide at a final concentration of 5% was included in the amplification of exon VIa. Exon VIb was amplified using primers 5‘-ATT GAT GGT TAG TAT TTT TTG TAG CAG TTG-3‘ (sense) and 5‘-TTA AAA ATA AAG TTA CAA AGG CCG GG-3‘ (antisense) with 30 cycles of 94°C/65°C/72°C for 30 seconds each.
The R195X and Y35X mutations were confirmed by restriction digestion with DdeI and RsaI (New England Biolabs, Queensland) respectively. The de novo SHOX R195X 583C>T mutation detected in family 9 has been previously reported.20 The IVS2-1G>A mutation was confirmed by PCR using the exon III antisense primer10 and sense primer 5‘- GCC CTG TGC CCT CCG CTC CAC -3‘, followed by digestion with BslI (New England Biolabs, Queensland).
PAR1 segregation analysis
Markers DXYS233 and DXYS23419 and the SHOX CA were amplified as previously described13 using fluorescent labelled primers, resolved on an ABI Prism 377 sequencer. To score the SHOX 792T>G SNP, the exon VIb PCR product was subjected to a further 25 PCR cycles of 95°C/65°C/72°C for 30 seconds each, using the nested primers 5‘- GTT TGC ACT TGG CCT TTT TTT TT-3‘ and 5‘- ACA TGG TCA AAC CCC ATC TCT GCT G-3‘. This amplicon was then digested with Fnu4HI (New England Biolabs, Queensland) and scored after electrophoresis. To score the SHOX 657A>G SNP, DNA sequencing of exon VIb was performed using the primers described earlier.10 To score the SHOX 63C>T SNP, PCR using the sense primer 5‘ TTT GAC CAG AAA AGC AAG GAC GGT AAC CG 3‘ and the exon II antisense primer from Rao et al10 was carried out. The amplicon was then digested with KspI (Roche, Sydney) and scored following electrophoresis.
SHOX reverse transcriptase PCR
Bone marrow fibroblasts (BMFs) were obtained from a direct bone marrow aspiration of the distal radius at the time of physiolysis. BMFs were grown by standard techniques in RPMI-1640 media supplemented with 10% heat inactivated fetal calf serum, penicillin (10 000 U/ml), and streptomycin (10 mg/ml) (Invitrogen, Melbourne). RNA was extracted using the RNeasy mini kit (Qiagen, Melbourne). RT-PCR reactions were carried out using a two step protocol. Reverse transcription was performed in 50 μl volumes containing 1-2 μg of total RNA, 3 ng random hexamer primers, 1 mmol/l each dNTP, 50 mmol/l Tris-HCl, pH 8.3, 75 mmol/l KCl, 1.5 mmol/l MgCl2, 10 mmol/l dithiothreitol, 10 mg bovine serum albumin, 100 units of RNaseOUT ribonuclease inhibitor (Invitrogen, Melbourne), and 400 units of Superscript II reverse transcriptase (Invitrogen, Melbourne). One to five μl of cDNA was used as template in each subsequent PCR. The RT-PCR product containing SHOX exons II to IV was amplified from cDNA as previously described.11 The SHOXb transcript was amplified with the same sense primer,11 and the exon VIb antisense primer used in genomic PCR above, using an annealing temperature of 59°C.
RESULTS
The cohort of 18 BMD patients comprised 16 females and two males with BMD. The pedigrees are depicted in fig 1. Nine of the 18 probands came from families in which dominant inheritance of Madelung deformity/mesomelic short stature was confirmed by assessment of other family members and/or strongly suspected in the earlier generations/unavailable relatives from anamnestic evidence. Apart from the father of the proband in family 5, all parents of probands were available for molecular testing in those families in which a SHOX mutation was identified. All probands had normal karyotypes. Thereafter, analysis by FISH, genomic sequencing, and linkage studies enabled the classification of BMD probands into a group with an identifiable SHOX mutation (families 1-9). A second set of pedigrees cosegregating LWD and a null allele at a SHOX intragenic/flanking polymorphic DNA marker (families 10-12) were identified by linkage analyses of SHOX intragenic/flanking markers. The third group consisted of probands (families 13-18) with no detectable mutation in SHOX. The mean height SDS of the 12 Madelung deformity probands with an identifiable SHOX aetiology was –2.0 (1.17) SDS and the remaining six probands (families 13-18) with wild type SHOX alleles was 0.0 (0.92) SDS (p=0.002).

Pedigrees of bilateral Madelung deformity probands.
FISH analyses identified large scale, hemizygous deletions of the SHOX locus in six of 18 probands, using the cosmid LLNOYCO3‘M‘34F5 (data not shown). These probands consisted of five females and one male (families 1-6). DNA sequencing of the SHOX coding region including exon VIb for the remaining 12 BMD probands identified three point mutations, including one de novo and two familial mutations. Two single nucleotide polymorphisms (SNP) were identified within exon VIb, including a silent polymorphism at nucleotide 657 (657A>G) and a second polymorphism within the 3‘ untranslated region of the SHOXb transcript (792T>G). Both of these polymorphisms were common in the general population with frequencies of 0.50 for 657A and 657G (n=32) and 0.64 for 792T (n=52). Another silent polymorphism at nucleotide 63, 63C>T, in exon II was identified in family 13, present in one of 44 control alleles (0.02).
Of the three point mutations, two introduced premature stop codons (Y35X, R195X) and the other (IVS2-1G>A) abolished the splice acceptor site at the boundary of intron 2 and exon III. A de novo SHOX R195X 583C>T mutation was detected in family 9 by DNA sequencing and was confirmed by restriction digestion with DdeI, as reported elsewhere.20 A nonsense mutation, Y35X, 105C>A in exon II, was detected in family 7. The cosegregation of this mutation with the LWD phenotype was tracked in this family by restriction digestion with RsaI. These results are shown in fig 2.
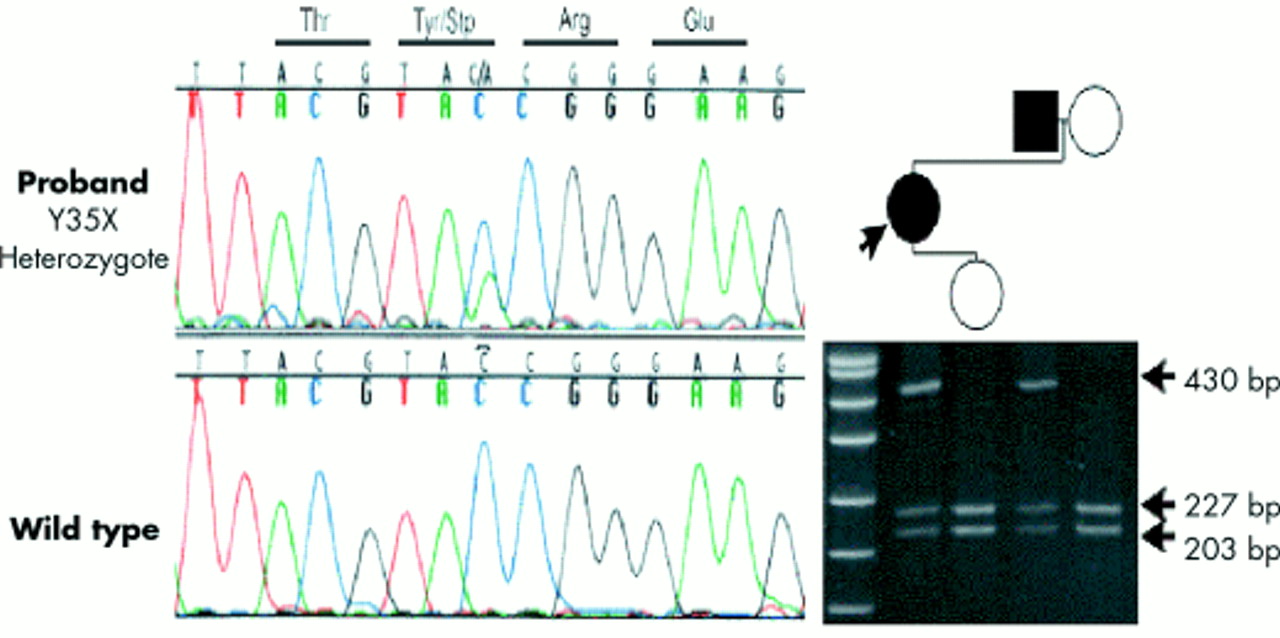
Detection of the Y35X mutation in family 7. In the left panel, DNA sequence of a portion of exon II shows coincidence of C and A at nucleotide 135 in the proband, and C in wild type. In the right panel, an acrylamide gel electrophoresis of the RsaI digested exon II PCR amplicon from members of family 7 shows coinheritance of the mutation and LWD. The size of the digestion products is indicated by the arrows. The left lane contains a DNA molecular weight marker, pUC19, digested with HpaII.
The IVS2-1 G>A mutation in the splice acceptor site at the boundary of intron 2 and exon III was identified in family 8. This mutation was shown to cosegregate with the LWD phenotype in this family and was not detected in 22 unaffected controls. These results are illustrated in fig 3A. If a cryptic splice site were activated as a result of this mutation, either skipping of exon III or retention of intron 2 would occur, thereby introducing downstream stop codons in the transcript from this allele. However, RNA extracted from BMFs of the proband, used for RT-PCR experiments, did not detect the presence of any aberrant SHOX transcripts. Genomic DNA sequencing established this proband to be heterozygous at the SHOX 792T>G SNP and subsequently RNA extracted from BMFs of the proband was used to evaluate the effect of the IVS2-1G>A mutation on SHOX transcription and splicing. Sequencing of RT-PCR products, specific for the SHOXb transcript confirmed that expression of the G allele only was present at the 5‘ SNP (SHOX 792T>G), as illustrated in fig 3B. Confirmation of this result was also obtained at nucleotide 657, where the proband’s genomic DNA was heterozygous (A/G) but sequencing of the SHOXb cDNA showed expression of the G allele only.

(A) Detection of the IVS2-1G>A mutation in family 8. In the left panel, DNA sequence of intron 2-exon III splice acceptor site shows both G and A, at the IVS2-1 nucleotide in the proband and only the G nucleotide in the wild type subject. In the right panel, the acrylamide gel electrophoresis of BslI digested PCR products in subjects from this family shows coinheritance of the mutation and LWD. The size of the digestion products is indicated by the arrows. The left lane contains the DNA molecular weight marker pUC19 digested with HpaII. (B) Demonstration of monoallelic SHOX expression because of the IVS2-1G>A mutation in the proband of family 8. The upper panels show the DNA sequence of PCR products derived from genomic DNA, which shows heterozygosity (T/G) at the SHOX 657A>G polymorphism and heterozygosity (A/G) at the SHOX 792T>G polymorphism. The lower panels show sequencing of the equivalent region in RT-PCR products amplified from bone marrow fibroblast RNA. This shows expression of a single SHOX allele, 792G, and 657A to be present.
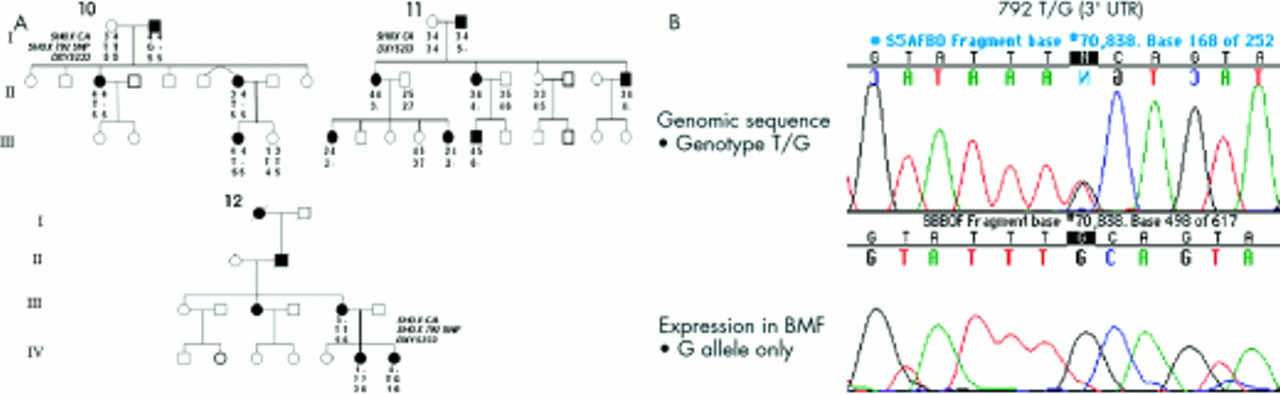
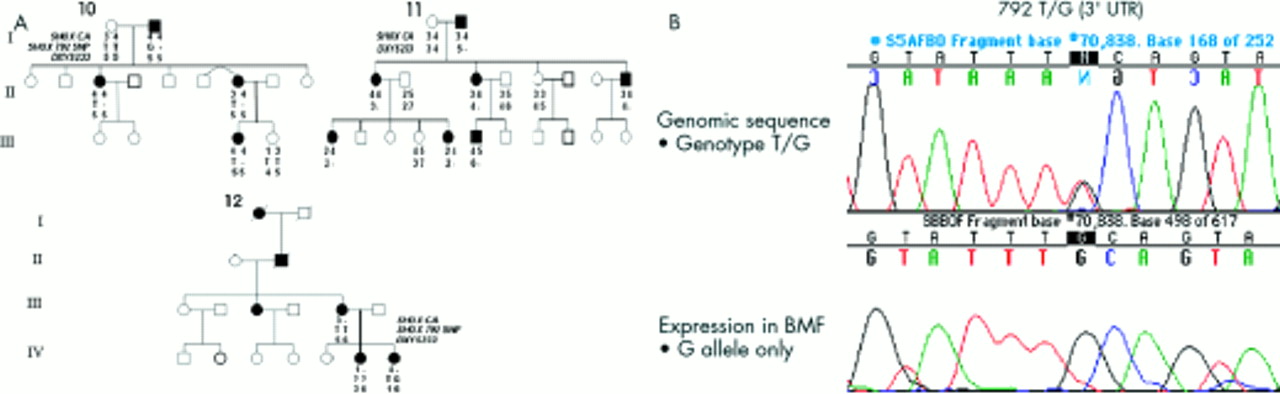
Linkage analyses of the SHOX intragenic CA repeat, the SHOX 792T>G SNP, and markers DXYS233 and DXYS234 were undertaken where possible, in those families in which no causative SHOX mutation had been identified. In three pedigrees (families 10-12), SHOX marker segregation analyses showed coinheritance of a null allele at a SHOX flanking/intragenic marker(s) and LWD. The results of these investigations are depicted in fig 4A. In family 11, the proband (III.6) coinherited BMD and a putative null allele at DXYS233. Total mRNA from BMFs obtained from III.6 at the time of physiolysis was used for reverse transcription and subsequent PCR and sequencing of SHOXb specific transcripts. Genomic DNA of III.6 was heterozygous for both the SHOX SNP at position 792 (T/G) and nucleotide 657 polymorphism (A/G). However, sequencing of the cDNA showed the presence of just the G allele at both positions, confirming monoallelic expression of the SHOXb transcript to be present in this subject (fig 4B, data not shown for the 657 polymorphism). In families 10 and 12, null alleles at the informative loci (SHOX 792 SNP and SHOX CA respectively) were also identified (fig 4A) but expression studies were unable to be performed, as BMFs were not available. In family 13, null alleles were not observed and linkage analyses were uninformative for families 14 and 15. Lack of parental samples for families 16-18 precluded such studies.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Segregation of the LWD phenotype and the SHOX polymorphic DNA markers (SHOX CA, the SHOX 792 SNP, and DXYS233) in families 10, 11, and 12. In family 10, I.2 had a null allele at the SHOX 792 SNP and no allele at this locus was inherited by either of his children, II.3 or II.6. III.3 was assumed to have a null allele from the presence of BMD. In family 11, I.2 had a null allele at locus DXYS233. No allele at this locus was inherited by his children II.1, II.2, or II.4. In addition, no allele at this locus was inherited by III.1 or III.5 from their mother II.1, or by III.6 from his mother II.2. In family 12, III.3 had a null allele at the SHOX CA locus. No allele at this locus was inherited by IV.6 or IV.7 from their mother, III.3. (B) Analysis of genomic DNA and cDNA from the proband III.6 in family 11. The top panel shows the results of DNA sequencing of genomic DNA. At nucleotide 792 in the 3‘ untranslated region of SHOXb, III.6 is heterozygous at this SNP, with T and G alleles. The lower panel shows the result of the sequencing of the cDNA obtained from RNA extracted from BMFs showing only the G allele at nucleotide 792 to be present, confirming monoallelic expression of SHOX.
DISCUSSION
In this study, an investigation of the molecular basis of Madelung deformity was undertaken in a surgical cohort of BMD patients, the majority of whom were female. Thereafter, identification of mutations in SHOX enabled LWD assignment in nine out of 18 BMD probands, and to a further three kindreds in which null SHOX flanking/intragenic alleles were coinherited with LWD. This results in a prevalence in this cohort of 67% of SHOX mutations in BMD probands, with only one familial case (family 13) not having a SHOX aetiology identified. Both familial (n=8) and de novo (n=3) instances of LWD were present. Although, our prevalence of de novo mutations of 33% (3/9) is higher than the 5% obtained from other series,9,13 our ascertainment differed from other studies, where familial cases were sought. Like other studies, we recorded female predominance.9,1321 In this study, six BMD probands (5/6 sporadic) were not identified by these analyses as having an identifiable mutation in SHOX. This finding is consistent with the results of other series and suggests that either the sensitivity of current molecular screening of SHOX is less than 100% and/or that mutations in SHOX regulatory sequences are present.21 Alternatively, genetic heterogeneity may exist for Madelung deformity, in part supported by the height discordance observed between the SHOX haploinsufficient probands (families 1-12) and the probands with SHOX wild type alleles (families 13-18).
The SHOX mutations encountered in this investigation encompassed six large scale hemizygous gene deletions detectable by FISH, of which two were de novo, and three point mutations identified by DNA sequencing of SHOX, of which one was de novo (R195X). Of the three SHOX point mutations identified in this investigation, two were novel while the R195X nonsense mutation had been previously reported in LWD12,20 and idiopathic short stature.10 Novel mutations included a nonsense mutation located in exon II, Y35X, and a splicing mutation, IVS2-1G>A. The Y35X nonsense mutation, if translated, would result in a severely truncated protein, lacking a homeodomain. The SHOX IVS2-1G>A mutation alters a nucleotide that is invariably conserved in the consensus splice acceptor sequence and is highly likely to interfere with normal splicing.22 Sequencing analysis of RT-PCR products obtained from BMFs showed that this particular mutation resulted in the loss of expression from one allele. This indicates that the mutant transcript is very likely to be rapidly degraded in vivo, as has been observed in other such splice site mutations.23,24
Segregation analyses using the SHOX CA repeat, the SHOX 792T>G SNP, and the DXYS233 dinucleotide repeat were undertaken. The physical order of these markers in distal Xp is telomere - SHOX CA - SHOX - DXYS233 - DXYS234 - centromere, determined from physical mapping of PAR1 (G Rappold, personal communication). This investigation identified putative null alleles for SHOX intragenic/flanking markers in three families (families 10-12), consistent with findings from another study of LWD.25 In family 11, the null allele at locus DXYS233 located 250 kb proximal to SHOX (G Rappold, personal communication) was evaluated for its effect on expression. Thereafter, RT-PCR of SHOX transcripts in BMFs from III.6 confirmed monoallelic expression of SHOX. Such null alleles may be associated with either an intragenic deletion of SHOX, or if confined to flanking markers a position effect may result in the silencing of SHOX transcription. Thus, in at least some instances, homozygosity for SHOX flanking/intragenic allele(s) is associated with monoalleleic expression and functional SHOX haploinsufficiency. Alternatively, a mutation at one of the primer binding sites or a deletion rearrangement involving one or both of the primer sites may be present. To date, the exact nature of the molecular pathology in the 5‘ or 3‘ region of SHOX that underlies such null alleles has not been elucidated.
By exploiting intragenic variation using SHOX SNPs, molecular confirmation of LWD was accomplished at the RNA level using DNA and RNA in this study. We examined lymphoblastoid cells lines for expression by RT-PCR, but no SHOX transcripts were detectable (data not shown), compatible with this being a rare RNA. SHOX expression has been observed from other tissues,10,11 but in reality only centres performing physiolysis will be able to obtain suitable tissue (BMFs) for this type of molecular analysis.
Acknowledgments
This work was supported by a PhD scholarship (SF), a RCHF Clinical Research Fellowship (CFJM), and the Variety Professorship support (JB), plus seeding grants from the Royal Children’s Hospital Foundation, Brisbane, Australia. In addition, further grant support was received from Novo Nordisc Pharmaceuticals, Australia (Grant in Aid), the Australian Paediatric Endocrine Group (CFJM), the Cressbrook Committee (CFJM), and Deutsche Forschungsgemeinschaft (GR and ER).