Article Text

Statistics from Altmetric.com
Editor—Although point mutations are responsible for many genetic disorders, it is increasingly being appreciated that many disorders can be caused by deletion or duplication of DNA sequences. Alterations in DNA copy number are responsible for several categories of disorders and can range in scale from a chromosome or chromosomal region to just one exon of a single gene. For instance, duplication and deletion of a 1.5 Mb DNA fragment on chromosome 17p11.2 containing the gene PMP22 is the common mechanism for Charcot-Marie-Tooth type 1A (CMT1A) and hereditary neuropathy with liability to pressure palsies (HNPP), respectively.1 At the level of individual exons, deletion of exon 172 or duplication of exon 133 of theBRCA1 gene can predispose to breast and ovarian cancer.
Cytogenetic studies (including CGH and array based CGH) can screen the whole genome for copy number changes. However, for most cytogenetic methods, the main disadvantage is low resolution, as they can detect deletions and amplifications only when several megabases of DNA are involved.4 Array based CGH may detect more localised changes,5 6 but at present is technically challenging and expensive to implement.
Gene dosage assessment at specific genetic loci is possible by using multiplex amplifiable probe hybridisation (MAPH),7 a simple, inexpensive, and non-microscopic approach to determining locus copy number in a complex genome. In this method, specific short probes flanked by the same primer pair are hybridised with a test genomic DNA sample. After stringent washing, these probes can be recovered and amplified quantitatively by PCR, and products resolved by polyacrylamide gel electrophoresis. The main advantages of this approach are high resolution (down to a few hundred base pairs) and the parallel processing of many DNA samples in a single experiment.
We have applied this method to genomic DNA from Holt-Oram syndrome (HOS, MIM 142900) patients in which the mutation atTBX5 was unknown. This syndrome8 9 is a rare developmental disorder (prevalence of about 1 in 100 000) with autosomal dominant inheritance, characterised by congenital heart malformations and upper limb defects. Mapped to 12q24.1, TBX5 is a member of the T box gene family, of which the protein products act as transcription factors via a conserved DNA binding domain (the “T domain”). Gene dosage alteration or target DNA binding derangement viaTBX5 mutations result in aberrant cardiogenesis.10 An overall mutation detection rate of 30% was reported by using SSCP (single stranded conformation polymorphism). More than 19 different non-translocation mutations inTBX5 have been shown to cause HOS.11 These consist of nonsense mutations, small deletions, rearrangements, insertions, and missense mutations. Some of these mutations introduce premature stop codons that are predicted to encode truncated polypeptides; these mutations appear to result in haploinsufficiency.9 Nevertheless, in many HOS pedigrees in which the disorder is linked to TBX5, a mutation has not been found. We therefore applied MAPH to look for large scale deletions or duplications atTBX5.
Methods
The TBX5 MAPH probe mixture consisted of three probes from the promoter region, 13 probes from the nine exons of the gene (exon 1, two probes; exon 5, two probes; exon 9, three probes; and other exons one probe each), and two probes 5 kb and 10 kb downstream of exon 9 (shown as “5K” and “10K”). As controls, there were six probes from other autosomes, one from the X chromosome, one from the Y chromosome, and also a non-human DNA probe. The aim of adding other autosomal chromosome probes was to allow the detection of whole TBX5 gene deletions or duplications. The non-human DNA probe was a control for specificity of hybridisation.
Probe preparation began with primer design to produce amplicons (differing by about 5-10 bp in size) between 140 and 600 bp; blunt ended amplicons were ligated into the EcoRV site of pZErO™-2 (InVitrogen) and then were transformed into E coli TOP10 (InVitrogen) by electroporation. Using flanking vector primers PZA (AGT AAC GGC CGC CAG TGT GCT G) and PZB (CGA GCG GCC GCC AGT GTG ATG), probes were amplified from plasmid DNA.
After denaturation by 1 μl 1 mol/l NaOH, human genomic DNA (1 μg) was immobilised on small pieces of nylon filter and then cross linked to the filters by UV irradiation (50 mJ). All filters were prehybridised with 1 ml prehybridisation solution (0.5 mol/l sodium phosphate, pH 7.2, 7% SDS, 1 mmol/l EDTA, and 100 μg/ml alkali denatured herring sperm DNA) at 65°C for more than two hours. Then, the prehybridised solution was replaced with 200 μl solution (from 300 μl prehybridisation solution plus 3 μl human Cot-1 DNA (Gibco BRL) boiled for two minutes) and incubated at 65°C for 30 minutes. Probe mixture (0.5 μl) was mixed with 1 μl (1 μg) human Cot-1 DNA, 2 μl (7 μg) E coli DNA digested with HaeIII, 2 μl (0.5 μg) ΦX174/HaeIII, and 2 μl “blocker mix” (20 μmol/l each PZAX (AGT AAC GGC CGC CAG TGT GCT GGA ATT CTG CAG AT)/PZBX (CGA GCG GCC GCC AGT GTG ATG GAT) primers). This mixture was denatured with 2 μl 1 mol/l NaOH and subsequently neutralised with 3 μl 1 mol/l NaH2PO4. The mixture was added to hybridisation solution and incubated at 65°C overnight. After hybridisation, the hybridisation solution was replaced with 1 ml prehybridisation solution, mixed, and removed. The filters were washed at 65°C for one hour sequentially with 500 ml 1 × SSC, 1% SDS (first 20 minutes) and 500 ml 0.1 × SSC, 0.1% SDS. Each filter was transferred to a 50 μl amplification reaction and the bound DNA amplified for five cycles (95°C for one minute/60°C for one minute/70°C for one minute) using primers PZA and PZB (stage 1 PCR). By using 32P 5′ end labelled primer PZA as one of the primers and 1 μl of (stage 1) PCR product as input, bound DNA was amplified (20 cycles, under the same conditions, followed by incubation at 72°C for 20 minutes to drive terminal dA addition to completion). Products were run on a denaturing 6% polyacrylamide/50% urea gel. Data captured by a storage phosphorimager screen (Molecular Dynamics) were quantitatively analysed with ImageQuant software.
Phosphorimager data were used to produce estimates of intensity for each band. The normalised ratio reflects the band intensity of a probe relative to its neighbours, which should be approximately constant for that probe across all the samples. To produce normalised ratios, each peak area (intensity of each band reported by IQ software) was divided by the sum of the two nearest autosomal control bands from the same sample. Then, the mean value of this ratio for each band was calculated across all samples (except that the Y chromosome probe intensity was divided by the number of male samples, and for X chromosome probes males were weighted as half of females). Finally, each ratio was normalised to the mean value, to give a simple numerical value (the “normalised ratio”) in which a value of 1.0 corresponds to diploid dosage for X linked and autosomal loci and to haploid dosage for Y linked probes.
Results
Analysis of TBX5 exonic copy number in genomic DNA from unaffected controls using MAPH shows reproducible results approximating to a normal distribution (data not shown). In the analysis of genomic DNA from 20 unrelated HOS patients (both sporadic and familial cases) with no known mutation inTBX5, we found one patient (fig 1) with a large deletion encompassing exons 3-9 (figs 2 and 3). This deletion was shared by the proband's father, but not detected in the proband's mother or his paternal grandparents. The normalised ratios of all probe signals in the deleted region were less than 0.75, and at least 3 SD below the normal mean (except H3, the third probe of exon 9) in the proband and his father. This was not seen in 21 out of 24 observations of promoter, exon 1, and autosomal control probes. For the probe from exon 2, intermediate values were observed (normalised ratios 0.76 and 0.63, 2.60, and 3.96 SD below the mean), suggesting the possibility that exon 2 is partially involved in the deletion. This deletion was not confirmed by other methods, but was reproducibly detected in replicate assays of DNA from the proband and his father.

Pedigree of the family studied in this report. DNA was analysed from the proband (35207), his affected father (24506), his mother (35198), and his paternal grandparents.

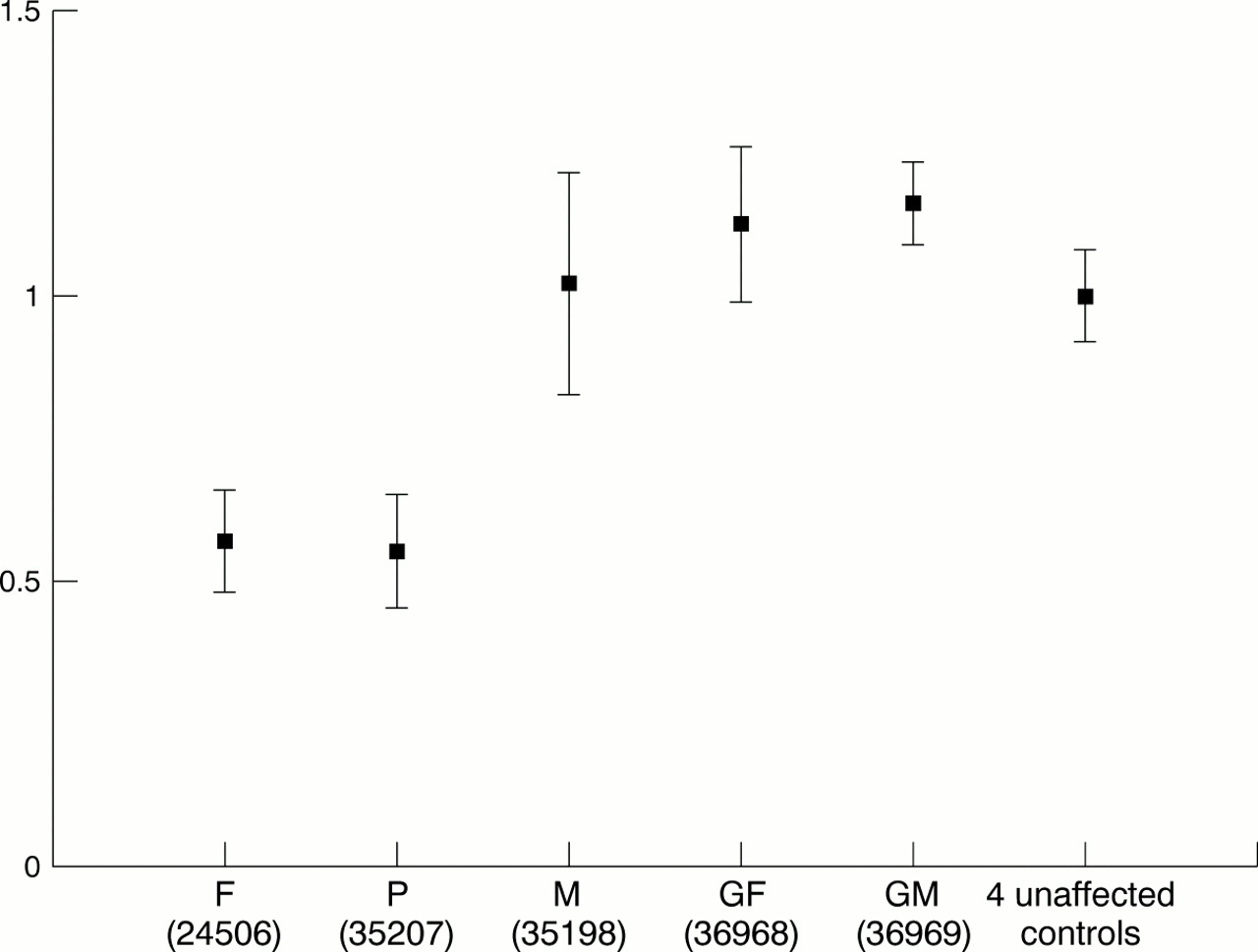
Mean of normalised ratio (error bar: 1 SD above and below the mean) of probes from the deleted region (exon 3-5K) in the proband and his parents and paternal grandparents, compared with the mean of four unrelated unaffected controls.

{kind=link}
{kind=link}
{kind=link}
Normalised ratio of probe from the TBX5 set in the proband and his father compared with the mean of four unrelated unaffected controls (error bar: 1 SD above and below the mean). The ratio of probes between exon 3-9 and 5K is about 0.5.
To confirm that the deletion in the proband's father was the result of de novo mutation, the identity of the DNA samples for the parents of 24506 (proband's father) was checked using hypervariable locus specific minisatellite probes MS205 (mapped to 16p13.3), MS620 (15q), and MS621 (5p). There was no evidence of any anomaly in the inheritance of alleles at these loci by individual 24506. Using conservative values (that is, upper estimates) for mean allele frequencies at MS205, MS620, and MS621 of about 3%,12 9%,13 and 2%,14 respectively, the combined probability of 24506's father having correctly sized alleles at all these three minisatellite loci purely by chance was estimated as approximately 4 × 10-4.
Using a series of primers at 1 kb intervals flanking the putative deletion end points (between exons 1 and 3 at the 5′ end and between probes 5K and 10K at the 3′ end), we attempted to amplify a fragment corresponding to the new junction at the deletion, but without success. This may indicate that the deletion event is complex, perhaps retaining some intronic material between the end points identified here.
This deletion could have been found using more standard quantitative PCR methods, but, in general, by screening all the exons of a gene in a single assay, MAPH has the advantage of complete screening of all exons for changes in copy number. Similarly, since the microsatellite AFMb023yd5 (D12S1646) maps to intron 7 ofTBX5, it would have been possible in principle to detect this deletion as anomalous segregation at this marker, though since this is the only intragenic microsatellite characterised at TBX5, events involving other parts of the gene would go undetected by such a screening strategy.
Overall, this was the only large deletion discovered in 20 unrelated HOS families examined; although the discovery of a deletion encompassing most of the gene strongly supports a haploinsufficiency mechanism in this kindred, it is clear that the majority of previously undiscovered mutations in TBX5 cannot be accounted for by large rearrangements. Consequently, our failure to find any exonic copy number change implies that some other explanation, such as promoter mutations, must be found for those families with HOS linked to 12q in which a TBX5 mutation has not been found.
The proband (35207) had multiple small apical ventricular septal defects (VSDs), which resolved spontaneously without treatment, and small thumbs, which he does not oppose very easily. The proband's father (24506) has bilateral thenar hypoplasia with poor extension of the thumbs. His heart is clinically normal now but he may have had a VSD that subsequently closed, since he was followed up for a heart murmur as a child. Unfortunately, further clinical details are not available. The paternal grandparents have normal hearts and limbs by history but have not been examined because they are not in the country.
Conclusions
In summary, Holt-Oram syndrome (HOS) is an autosomal dominant disorder, characterised by congenital heart and upper limb malformations. Although genetically heterogeneous, HOS is frequently linked to the gene TBX5, in which mutations have been detected in affected subjects. In many patients with HOS, however, conventional methods have failed to show a mutation inTBX5, suggesting that some of the “missing” mutations may be due to exonic deletions. We applied multiplex amplifiable probe hybridisation (MAPH) to genomic DNA from patients in which the mutation at TBX5 was unknown. We report a large deletion encompassing exons 3-9 in two related patients. Although the causative mutation is still unknown in many HOS patients, this observation supports a haploinsufficiency mechanism for HOS.
Acknowledgments
We appreciate the help of Dr Zeinali (Iran Pasteur Institute), Dr Bernadette Farren, and Stephen Cross in this study. We thank the Islamic Republic of Iran Health Ministry, the Wellcome Trust, and the British Heart Foundation for their support.