Article Text

Statistics from Altmetric.com
Editor—Cri du chat syndrome (CdCS) is one of the more common deletion syndromes, involving the short arm of chromosome 5, with an incidence of 1 in 50 000 live births. Classically, patients with this syndrome present with microcephaly, a round face, hypertelorism, micrognathia, prominent nasal bridge, epicanthic folds, hypotonia, and severe psychomotor retardation. Infants also exhibit a high pitched cry similar to the mewing of a cat, which is usually considered diagnostic for this syndrome.1 2 Recently, genotype-phenotype studies in CdCS led to the identification of two separate chromosomal regions, hemizygosity for which is associated with specific phenotypes.3 A deletion of 5p15.3 results in the manifestation of a cat-like cry4, while a deletion of 5p15.2 results in the presentation of the other major clinical features of the syndrome.5 Moreover, a region for speech delay in 5p15.3 has been identified.6
From a review of 331 published cases, Niebuhr2 estimated that most CdCS cases are the result of de novo deletions (about 80%), some derive from a familial rearrangement (12%), and only a few show other rare cytogenetic aberrations, such as mosaicism (3%), rings (2.4%), and de novo translocations (3%). Chromosomal mosaicism in CdCS has been described involving a cell line with a 5p deletion and a cell line with a normal karyotype.2 7
We describe the first three reported cases of mosaic de novo 5p anomalies involving two rearranged cell lines in CdCS out of 80 (3.75%) patients from the Italian Register of CdCS8analysed in a large study of correlation between 5p deletion and phenotypic effect (Cerruti Mainardi et al, manuscript in preparation).
Patient 1 was the first female child of healthy, unrelated parents. At birth the mother was 29 and the father 31 years old. The pregnancy was complicated by a threatened miscarriage in the 12th week of gestation. There had been no exposure to alcohol, nicotine, drugs,x rays, or teratogens during pregnancy. Her younger sister was healthy. The family history was unremarkable.
The baby was born by caesarean section because of reduced uterine contractions in the 39th week of gestation. Apgar scores were 7 and 6 at one and five minutes, respectively. Her birth weight was 2750 g (10th-25th centile), length was 48 cm (25th centile), and head circumference was 34 cm (25th-50th centile).
The neonatal course was complicated by transient respiratory distress and metabolic acidosis. In the first days of life, sucking difficulties were reported, requiring nasogastric feeding. Neonatal physical examination showed epicanthic folds and hypertelorism. CdCS was suspected because of an abnormal cry and was confirmed by cytogenetic analysis. No significant medical illness was observed during infancy.
On physical examination at the age of 6 years, weight was 20.7 kg (50th centile), height was 114.5 cm (50th centile), and head circumference was 49 cm (3rd-10th centile). Clinical features included a high voice, downward slanting palpebral fissures, broad nasal bridge, hypertelorism, epicanthic folds, slight micrognathia, slightly low set ears, diastasis recti, and flat arches of the feet. Dysmorphism was felt to be mild (fig 1A).

(A) Case 1 aged 6 years. (B) Ideogram of the normal and the abnormal chromosomes 5 in both cell lines. Specific single locus probes used to define breakpoints are shown. (C) FISH performed with D5S778 lambda phage probe shows enhanced medial signal strength on one chromosome 5 consistent with a duplication.
However, developmental delay, evaluated with the Denver Developmental Screening Tests,9 was present: she sat with her head steady at 6 months, sat without support at 10 months, and walked well at 2 years. Speech delay was more severe; she spoke her first words at 4 years and could combine two different words at 6 years.
Chromosome analysis of the patient, performed on peripheral blood lymphocytes, showed two cell lines; in 73% of cells a 5p deletion was observed and in 27% of cells a 5p duplication was observed.
FISH analysis performed with specific single locus DNA lambda phage probes spanning 5p10 identified breakpoints both on the deleted chromosome 5 and on the duplicated one. The breakpoint on the deleted chromosome 5 is in p14.1 between D5S769 and D5S711 and the breakpoints on the duplicated chromosome 5 are in p15.2 between D5S778 and D5S751 and in p12 between D5S802 and the centromere. The duplication is inverted.
The cytogenetic and FISH results are summarised in fig 1B and C. In conclusion the karyotype was interpreted as: 46,XX,ish del(5)(:p14.1→qter)(D5S711-)[73]/46,XX,ish dup(5)(:p12→p15.2::p15.2→qter)(D5S802++, D5S778 ++, D5S751-)[27]. The parents' and sister's karyotypes were normal.
Molecular analysis performed with microsatellite D5S211411indicated the paternal origin of the deleted and the duplicated chromosomes 5.
Patient 2 was the first female child of healthy, unrelated parents. At birth the mother was 26 and the father 31. Pregnancy was complicated by bleeding in the first 12 weeks; ultrasound examination showed intrauterine growth retardation. There was exposure to nicotine during pregnancy (15 cigarettes a day). The family history was remarkable for two deaf maternal aunts.
The baby was delivered in the 40th week of gestation. Her birth weight was 2680 g (3rd-10th centile); length and head circumference were not recorded. The neonatal course was complicated by metabolic acidosis. Cytogenetic analysis, performed in another laboratory because of dysmorphism, showed a karyotype with three cell lines: 46,XX,del(5) (p15)[65]/46,XX[33]/46,XX,dup(5)(p13→pter) [2].
During infancy, she suffered from recurrent bronchial and pulmonary infections requiring admission to hospital. There was an episode of seizures and psychomotor development, evaluated with Denver Developmental Screening Tests,9 was delayed; she was able to sit at 1 year and to walk and speak her first words at 4 years.
Physical examination at 5 years showed weight 18.5 kg (50th centile), height 104 cm (10th-25th centile), and head circumference 47 cm (<3rd centile). Clinical features included a high voice, broad nasal bridge, hypertelorism, epicanthic folds, myopia and mild convergent strabismus, downturned corners of the mouth, micrognathia, low set ears, left preauricular tag, transverse flexion creases, clinodactyly, diastasis recti, flat arches of the feet, and anteriorly placed anus (fig 2A). She showed severe mental and speech delay and hyperactive behaviour.

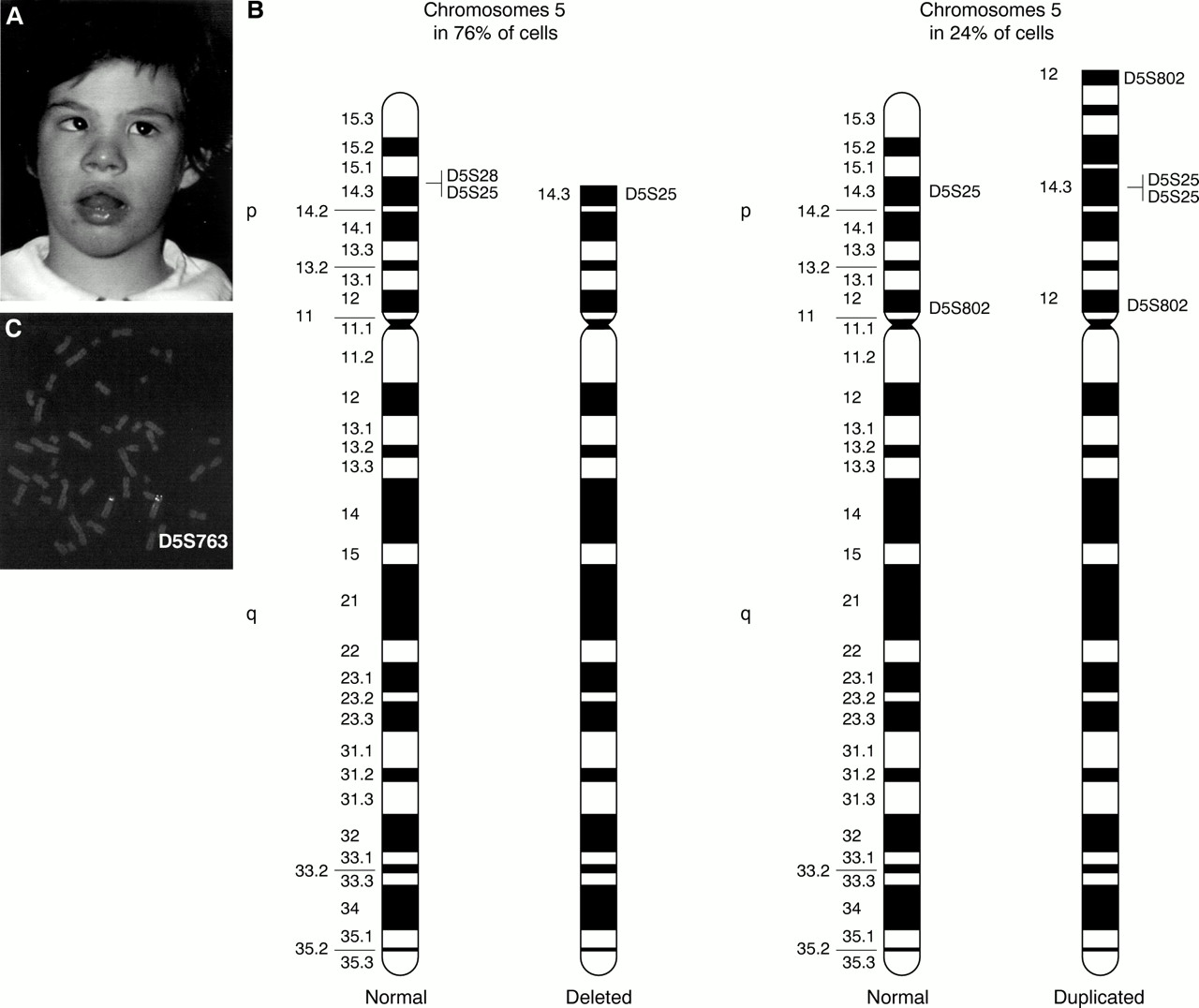
(A) Case 2 at 5 years. (B) Ideogram of the normal and the abnormal chromosomes 5 in both cell lines. Specific single locus probes used to define breakpoints are shown. (C) FISH performed with D5S763 lambda phage probe shows a duplication on one chromosome 5.
Chromosome analysis of the patient, performed on peripheral blood lymphocytes, showed two cell lines; in 76% of cells a 5p deletion was observed and in 24% of cells a chromosome 5 with normal dimensions and banding pattern was observed.
FISH analysis performed with specific single locus DNA lambda phage probes spanning 5p10 established that the apparently normal chromosome 5 really represented a 5p duplication and identified breakpoints both on the deleted and the duplicated chromosomes 5. The breakpoint on the deleted chromosome 5 is in p14.3 between D5S25 and D5S28 and the breakpoints on the duplicated chromosome 5 are in p14.3 between D5S25 and D5S28 and in p12 between D5S802 and the centromere. The duplication is inverted.
The cytogenetic and FISH results are summarised in fig 2B and C. In conclusion the karyotype was interpreted as: 46,XX, ish del(5)(:p14.3→qter)(D5S28−)[76]/46,XX,ish dup(5)(:p12→p14.3::p14.3→qter)(D5S802++,D5S25++, D5S28−)[24]. The parents' karyotypes were normal.
Molecular analysis performed with microsatellite D5S198111indicated the paternal origin of the deleted chromosome 5.
Patient 3 was the first female child of healthy, unrelated parents. At birth the mother was 30 and the father 33. The pregnancy was uneventful and there was no exposure to alcohol, nicotine, drugs,x rays, or teratogens during pregnancy. Her younger sister and brother were healthy. The family history was unremarkable.
The baby was delivered at 40 weeks of gestation. Her birth weight was 2600 g (3rd -10th centile), length was 50 cm (50th-75th centile), and head circumference was 33 cm (10th-25th centile). CdCS was suspected because of dysmorphism, microcephaly, and a high pitched monotonous cry and was confirmed by cytogenetic analysis. Talipes of the left foot was observed.
At the ages of 3½ and 5½ she underwent two operations for left cholesteatoma. During infancy her psychomotor development, evaluated with Denver Developmental Screening Tests,9 was delayed; she was able to grasp objects at 8 months, to sit at 2 years, to say her first words at 3 years, and to walk at 3½ years. Menarche occurred at 11 years.
On physical examination at 12 years, her weight was 40 kg (50th centile), height was 137 cm (<3rd centile), and head circumference was 48 cm (<3rd centile). Clinical features included a high voice, epicanthic folds, divergent strabismus, micrognathia, a high arched palate, slightly low set ears, incomplete bilateral flexion creases, diastasis recti, flat arches of the feet, and talipes of the left foot (fig 3A). She showed severe mental and speech delay and hyperactive behaviour.
{kind=link}
{kind=link}
{kind=link}
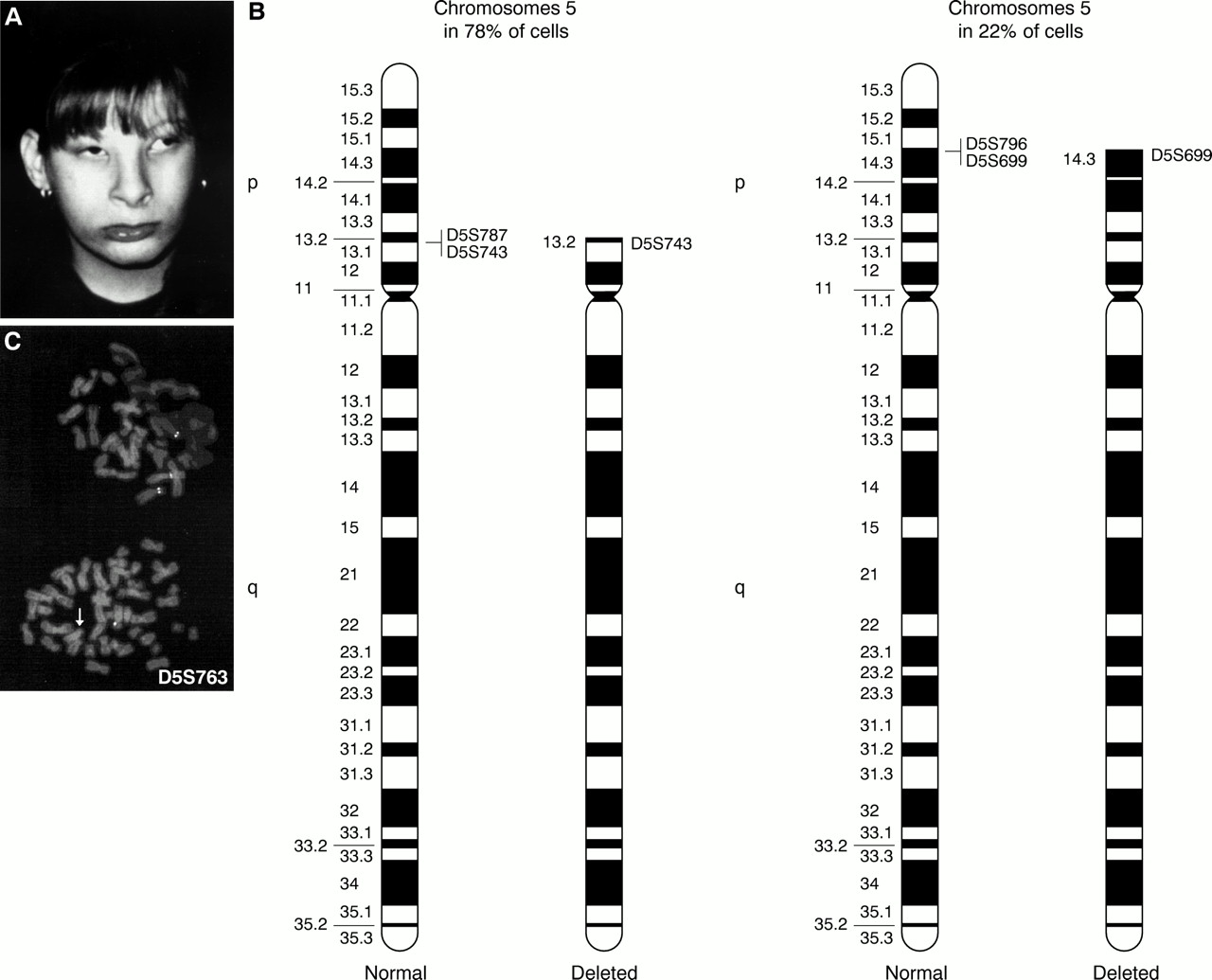
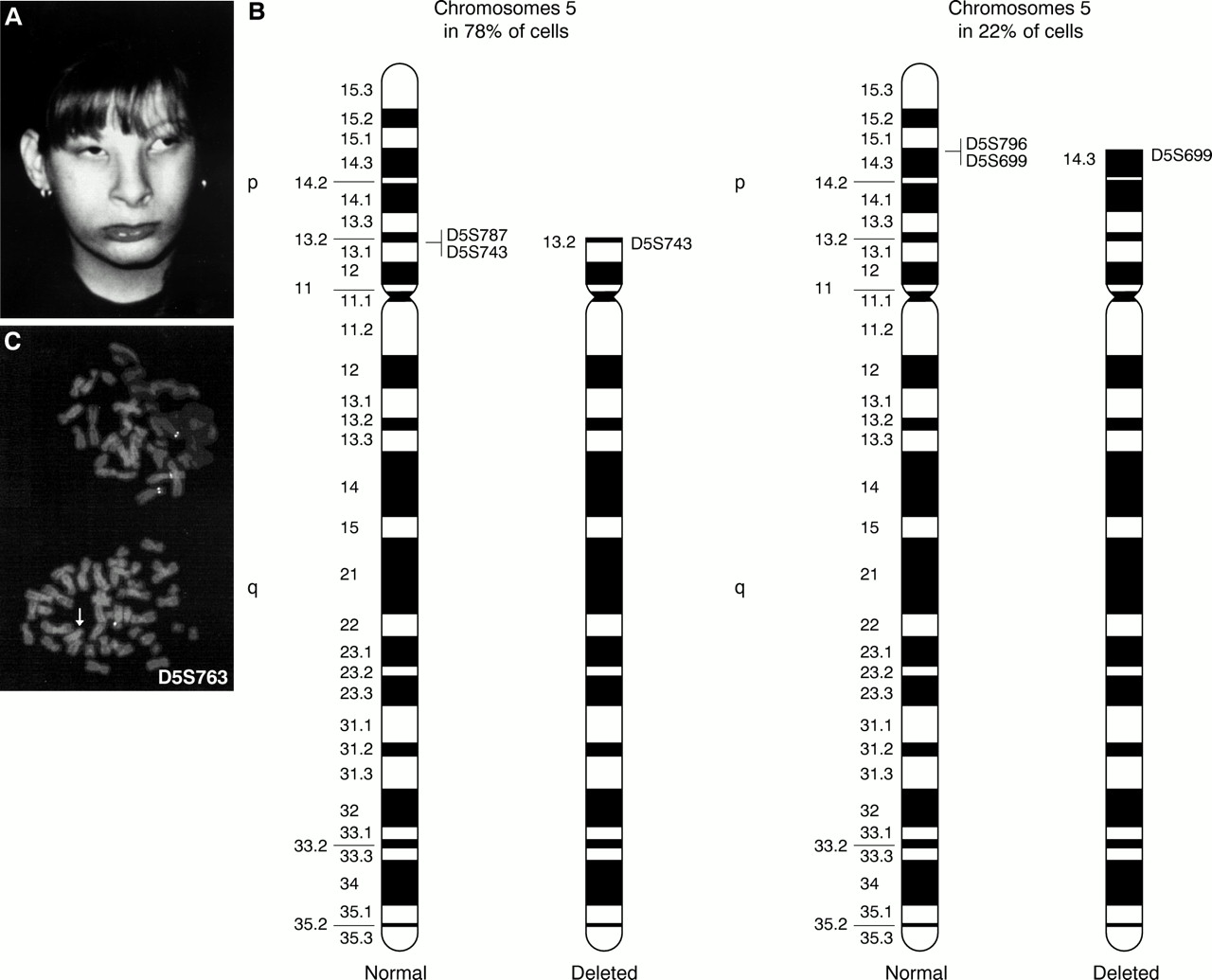
(A) Case 3 at 12 years. (B) Ideogram of the normal and the abnormal chromosomes 5 in both cell lines. Specific single locus probes used to define breakpoints are reported. (C) FISH performed with D5S763 lambda phage probe shows two different deletions in the two adjacent metaphases; in the upper one the probe is not deleted, while in the lower it is deleted (arrow).
Chromosome analysis of the patient, performed on peripheral blood lymphocytes, showed two cell lines with different 5p deletions: in 78% of cells the deletion extended from pter to p13 and in 22% of cells the deletion extended from pter to p14.3.
FISH analysis performed with specific single locus DNA lambda phage probes spanning 5p10 allowed finer definition of the extent of the deletions. The breakpoint on the more deleted chromosome 5 is in p13 between D5S743 and D5S787 and the breakpoint on the less deleted chromosome 5 is in p14.3 between D5S699 and D5S796. The cytogenetic and FISH results are summarised in fig 3B and C.
In conclusion, the karyotype was interpreted as: 46,XX,ish del(5)(:p13→qter)(D5S787−)[78]/46,XX,ish del(5)(:p14.3→qter)(D5S796−)[22]. The parents' karyotypes were normal. Molecular analysis performed with microsatellite D5S198711 indicated the maternal origin of the deleted chromosome 5.
In summary, as a consequence of these rearrangements of chromosome 5p, at least in peripheral blood lymphocytes, patients 1 and 2 have a partial monosomic cell line and a partial monosomic/trisomic cell line, while patient 3 shows two different partial monosomies.
It is interesting to analyse the CdCS phenotype of patients 1 and 2 in relation to the presence of partial trisomy 5p in a proportion of cells. Partial trisomy 5p was first described in 1964,12but it is a very rare event. Most partial trisomies 5p are the consequence of an unbalanced translocation with another autosome. However, although some manifestations may differ from case to case because of the effect of the monosomy of the other chromosome involved in the translocation, a clinically recognisable and consistent phenotype is always present and is apparently little influenced by the presence of concomitant autosomal imbalance. The most common manifestations are normal birth weight, macrodolichocephaly, downward slanting palpebral fissures, epicanthus, bulbous nose, low set ears, short big toes, hypotonia, seizures, and psychomotor retardation. Multiple congenital anomalies are present and early death is frequent.13-19 The extent of the clinical findings in the reported cases depends on the length of the trisomic portion. The larger the trisomy, the more pronounced are the clinical signs. Physical signs are almost absent when 5p14-ter is involved, while those cases with trisomies of chromosome material including 5p13 or the complete short arm have more severe multiple congenital anomalies, mental retardation, and growth failure.20 Signs and malformations described in patients with partial monosomy 5p and in patients with partial trisomy 5p compared to those observed in patients 1 and 2 are summarised in table 1.
Of particular interest are the mild phenotype in patient 1 and the comparison between patients 1 and 2 (figs 1A and 2A). From the developmental point of view, the two patients are comparable since they are approximately the same age, the parents' educational levels are similar, and they followed the same therapeutic programme with the same person. Patient 1 shows a CdCS phenotype with the cat-like cry, dysmorphism, and developmental and speech delay. However, her phenotype is very mild in relation to the deletion breakpoint in 5p14.1 (D5S711), especially when compared with the results obtained from a wider study on CdCS (Cerruti Mainardi et al, manuscript in preparation). The large duplication of 5p12-5p15.2 apparently does not produce any significant phenotypic effect. This is contrast to patients with duplication of 5p. Patient 2 shows a severe CdCS phenotype in relation to the deletion breakpoint in 5p14.3 (D5S28). Some clinical features and malformations are attributable to partial trisomy 5p and others are common both to partial monosomy and partial trisomy 5p (table 1).
The phenotype of patient 1 is milder than patient 2, although the deletion is bigger. We therefore speculate that in patient 1, 5p deletion and 5p duplication could compensate for each other with the duplicated cell line contributing a double dosage of the critical region in 5p15.2,5 while the cat-like cry4and the speech delay regions6 are present in single dose (fig 1B). This compensation is not possible in patient 2, where the region between 5p14.3 and the telomere is deleted in both cell lines (fig 2B). Overall, in both cases the CdCS phenotype prevails.
The cell line with the deletion is more prevalent compared with that with the duplication, at least in peripheral blood lymphocytes. This would suggest that the deletion has been favoured during development because monosomy 5p is less severe than trisomy 5p, as supported by the fact that monosomy 5p is far more common than trisomy 5p.
Patient 3, where both cell lines have a deletion (fig 3B), shows a classical CdCS phenotype (fig 3A) and its severity could be because of the prevalence of the more deleted cell line.
We report a frequency of mosaicism in CdCS of 3.75%. To the best of our knowledge, this is the largest record of cases that allows an estimation of frequency from a homogeneous pool of data. Mosaicism for structural aberrations is generally thought to be very uncommon, and the relative frequencies of the various types of structural aberrations, including deletions, have not been established. Data available on chromosomal structural rearrangements report a frequency of mosaicism varying from 0.008% in a postnatal study to 0.09% in a prenatal study.21 22 Hence, the frequency of mosaicism in CdCS appears to be very high if compared with other chromosomal rearrangements. Many hypotheses could explain this observation. The real frequency of mosaicism in chromosomal structural rearrangements could be understimated either because of the small number of metaphases analysed or because most studies were performed using only cytogenetic analysis and some subtle rearrangements could have been missed. For instance, in patient 2 the duplicated chromosome 5 was clearly distinguishable only by the use of molecular cyotgenetics with locus specific probes. The instability of the short arm of chromosome 5 could be attributable to the unusual richness of low or medium copy number repetitive sequences, such as LINE-1 elements, chromosome 5 specific repeats, or expressed pseudogenes,23-25 causing aberrant recombination and rearrangements within the region. There is increasing evidence that chromosome rearrangements resulting from unequal crossing overs between homologues, unequal sister chromatid exchanges, meiotic recombination within a single chromatid, and excision of intrachromatid loops may result from the presence of repeated sequences in the human genome.26-28
The absence of a normal cell line in all three cases presented here is consistent with a prezygotic origin of inverted tandem duplication. Different mechanisms have been proposed to explain the origin of such duplications.29-35 In these cases, the explanation is complicated by the presence of mosaicism and by the absence of preferential breakpoints. We could postulate that, in all cases, the first common event is the formation of a dicentric chromosome by recombination between mispaired copies of repeated and inversely oriented sequences at meiosis I; then, at anaphase I, a breakage of the resulting dicentric isochromosome would cause, respectively, the inv dup(5p) in patients 1 and 2 and a del(5p) in patient 3. Mosaicism could then arise mitotically at the level of a single DNA strand where loop formation and unequal crossing over between repeated sequences could cause breakage of the strand at different points and generate the deleted cell line in patients 1 and 2 and the line with a bigger deletion in patient 3.
In conclusion, these patients represent the first three cases of de novo 5p anomalies involving two rearranged cell lines, and provide new insights into mosaic structural rearrangements and into genotype-phenotype correlations in CdCS.
Acknowledgments
The first two authors contributed equally to this work. We thank the families for their cooperation and Mr Domenico Carratta for his skilful photographic assistance. This work was supported by an Italian Telethon Foundation grant (E.511) and by the Italian Cri du Chat Children Association (ABC). IL-2 lymphocyte cell lines of patients are stored at the Galliera Genetic Bank, supported by an Italian Telethon Foundation grant (C.42).