Article Text

Statistics from Altmetric.com
Editor—Wolf-Hirschhorn syndrome (WHS), first described independently in 1965 by Wolf et al 1 and Hirschhorn et al,2 is a well defined multiple congenital anomalies/mental retardation syndrome resulting from deletion involving chromosomal band 4p16.3 with a minimal critical region of 165 kb.3
Different mechanisms are responsible for WHS, including terminal 4p deletions, familial translocations, and de novo complex chromosomal rearrangements such as unbalanced translocations. The frequency of translocations in WHS as a result of parental chromosomal translocations is estimated to be 5 to 13%,4 5 whereas the rate of sporadic translocations in WHS was suggested to be 1.6% (2/120).5 A few isolated cases with de novo translocations5-12 resulting in WHS have been described recently. The trisomic segment, however, often could not be defined owing to the lack of specific cytogenetic techniques.5 6 8 13-15
Here we report six patients with unbalanced translocations, t(4p;8p) and t(4p;7p), respectively, and discuss their phenotypic abnormalities.
Since 1996 we have performed clinical, molecular, cytogenetic, and molecular-cytogenetic investigations in a total of 22 patients with clinical signs of Wolf-Hirschhorn syndrome. These patients were all seen by one of the authors. In five of them (22%), the combination of cytogenetic and molecular investigations showed a de novo unbalanced translocation; in a sixth patient the de novo occurrence of the translocation could not be confirmed because no blood sample from the father was available.
The clinical findings of patients 1-5, who all presented with characteristic features of WHS, are summarised in table 1. Patient 6 (CL220585/87E1624) had some additional findings listed in table1.
Clinical findings in patients 1-5 with t(4;8) and patient 6 with t(4;7)
The combination of Hirschsprung disease and hydrocephalus was suggestive of CRASH syndrome in patient 6, but SSCP analysis of theL1CAM gene was normal.
Chromosome studies including GTG banding were performed on peripheral blood lymphocytes according to slightly modified standard techniques.16
DNA from cosmids pC847.351,17 L21f12 (D4S180), and L228a7 (D4S81)18 and inter-Alu PCR products19from YAC 877G6, 405D10, 794D12, 225D2, and 435A11 (CEPH) were labelled with digoxigenin using a BRL nick translation kit (Gibco, Life Technologies Inc, Gaithersburg, MD, USA). Labelled cosmid and YAC DNA was separated from unincorporated nucleotides by using 1800 ml Centricon 30 (Amicon Inc, Beverly, MA 01915, USA) filters. FISH was performed as described by Lichter et al.20 Labelled cosmid DNA (50 ng) or YAC DNA (100 ng) was mixed with human Cot-1 fraction DNA to suppress repetitive sequences or block non-specific hybridisation. Hybridisation was detected using monoclonal anti-digoxin, anti-mouse IgG FITC conjugate and anti-rabbit IgG FITC conjugate (Sigma Immuno Chemicals, St Louis, USA) and counterstained using propidium iodide or DAPI.
A cosmid derived from D4S96, the 8p telomere probe and 7p distal probe (Oncor, Gaithersburg, VA, USA) and the whole chromosome paints 4 and 8 (AGS, Heidelberg, Germany/Oncor, Gaithersburg, VA, USA) were hybridised according to the manufacturer's instructions. For the hybridisations, clone pGXba11/34021 was used as centromeric control for human chromosome 4.
Genomic DNA was prepared from peripheral blood lymphocytes or EBV transformed lymphoblastoid cell lines from patients and their parents according to previously described techniques.22 23
Genotyping of chromosome 4p was performed using the following previously described microsatellite markers: D4S1182 (Acc ID GDB 19723924), D4S43 (Acc ID GDB 12436024), ADRA2C (Acc ID GDB 37083425), HOX7 (Acc ID GDB 15699126 27), D4S403 (Acc ID GDB 18810628), D4S2366 (Acc ID GDB 684453), D4S2639 (Acc ID GDB 685881), and D4S2397 (Acc ID GDB 683982). A new microsatellite, 75B9Rep, was generated by one of the authors based on sequence information of cosmid 75B9A (Acc No Z69651) which is derived from the distal end of the Huntington's disease cosmid contig.29 Oligomers were designed from sequences flanking the compound dinucleotide repeat (CT)2G(CT)26(CA)14. The following primer sequences were used for the amplification of the repeat, forward primer 5′ CTGAACTTTATCCAATTAGTCTTG 3′ and reverse primer 5′ GAATCTTTCTGTCCCACGAT 3′. This primer set amplifies a 227 bp DNA fragment product. PCR reactions were performed in 10 μl volumes containing 100 ng DNA, 0.5 U GenecraftR Taq polymerase, 1 × GenecraftRPCR buffer, 2 μmol/l dNTP, 4 pmol/l of each primer, 1 μCi32P-dCTP (3000 Ci/mmol/lol, Amersham), and 2 mmol/l MgCl2. Formamide (2%) was added to increase stringency of the reaction. After an initial denaturation of one minute, 30 cycles of one minute at 94°C, 30 seconds at 57°C, and 30 seconds at 72°C were carried out in a Crocodile IIIR (Appligene) thermocycler. The PCR products were resolved on 6% polyacrylamide gels flanked by a sequence ladder (M13, −40) as size marker. The Hu4/Hu5 (Acc ID GDB 249651) primer pair amplifies the CAG repeat of the huntingtin gene. Primer Hu4 has been published by Riesset al 25 (primer sequence: Hu4 5′ ATGGCGACCCTGGAAAAGCTGATGAA 3′), and primer Hu5 has been published by Rubinsztein et al 30 (primer sequence: Hu5 5′ GGCGGTGGCGGCTGTTGCTGCTGCTGC 3′).
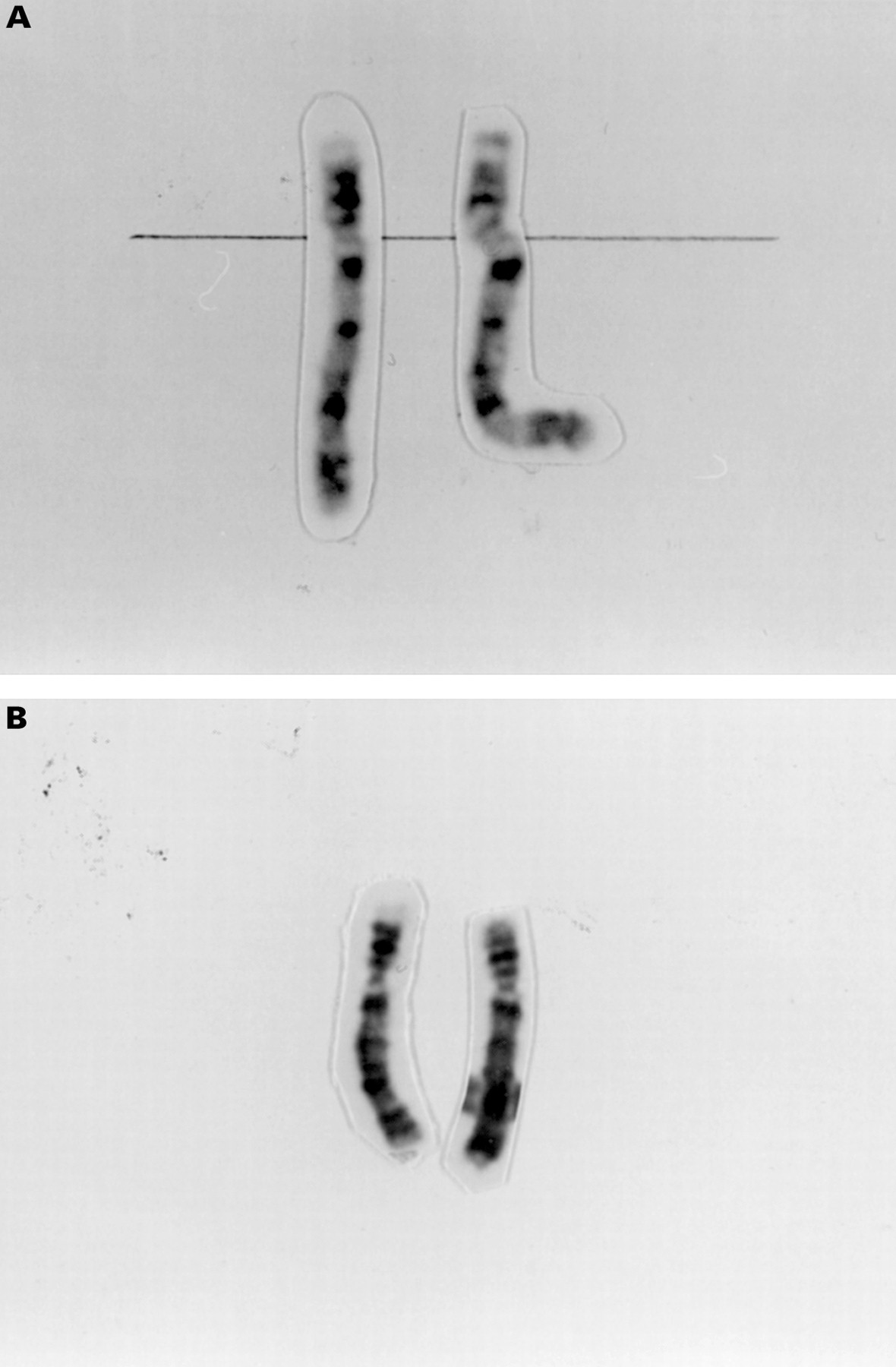
At the beginning of our study of 22 WHS patients, we reanalysed the chromosomes in lymphocytes by standard Giemsa banding techniques. At a banding resolution of at least 450 bands per haploid genome,31 an additional small G positive band was observed on the distal short arm of one chromosome 4 in patients 1 and 2 (fig1A) and an unbalanced translocation was suspected in patients 3, 4, and 5 (fig 1B). In patient 6, a deletion of 4p with breakpoints in 4p16.2 was suggested. Thus, we suspected an unbalanced translocation between the short arm of one of the chromosomes 4 and another unidentified chromosome in patients 1-5, while the cytogenetic aberration in patient 6 appeared to be a simple deletion.

(A) Chromosomes 4 of patient 2 showing an additional G positive band in 4pter. (B) Chromosomes 4 of patient 4 showing same length of 4p but different G banded pattern.
Chromosomal analysis in all participating parents showed a normal karyotype. In patient 5 the father was not available. The remaining 16 patients of this study are described elsewhere (D Wieczorek, manuscript in preparation).
Different cosmids were used to confirm the clinical diagnosis of WHS in all patients. In patients 1, 2, and 5, a deletion of D4S96 (Oncor) and of cosmids pC847.351 and L228a7 on one homologue of chromosome 4 was noted (fig 2A). In patients 3 and 4, YAC 405D10 was also deleted on one homologue. In patient 5, the size of the deletion was determined by FISH only. Cosmids pC847.351, L21f12, and L228a7 and YACs 877G6 and 405D10 were deleted on one chromosome 4, whereas YACs 794D12, 225D2, and 435A11 were present on both chromosomes 4 (table2).

FISH results in patient 4. (A) FISH with cosmid pC847.351 showing lack of signal in one chromosome 4p. (B) FISH with wcp(4) showing fluorescent signals on the entire length of both chromosomes 4 except for the terminal region of one chromosome 4. (C) FISH with distal 8p probe showing two signals in the distal region of 8p and an additional signal in the distal short arm of one chromosome 4.
Extent and origin of the 4p deletion in patients 1–6
FISH staining with a whole chromosome 4 paint (AGS/Oncor) in all six patients gave hybridisation signals along the entire length of both chromosomes 4 except for the distal part of the aberrant chromosome 4p (fig 2B). Both chromosomes 8 showed hybridisation signals along the entire length with whole chromosome paint 8 (AGS), but also on one chromosome 4 in patients 1-5. FISH with an 8p telomere probe (Oncor, Gaithersburg, VA, USA) showed signals on both chromosomes 8 and an additional signal on one chromosome 4 (fig 2C).
Based on these findings we determined the karyotype of patients 1 and 2 as 46,XX,der(4),t(4;8)(p16.3;p23.1).ish t(4;8)(wcp4−, wcp8+, 8ptel+, pC847.351−, D4S96−, 228a7−; wcp4−, wcp8+, 8ptel+) and in patients 3, 4, and 5 as 46,XX,der(4),t(4;8)(p16.2;p21).ish t(4;8)(wcp4−, wcp8+, 8ptel+, pC847.351−, D4S96−, 228a7−, 405D10−; wcp4−, wcp8+, 8ptel+).
FISH was also performed in the parents of patients 1-4 and the mother of patient 5. Normal results were detected with D4S96 (Oncor) and with the 8p telomere probe (Oncor). We conclude that the translocation t(4;8) occurred de novo in all patients. However, in patient 4 one could not exclude that the translocation may be inherited from the father.
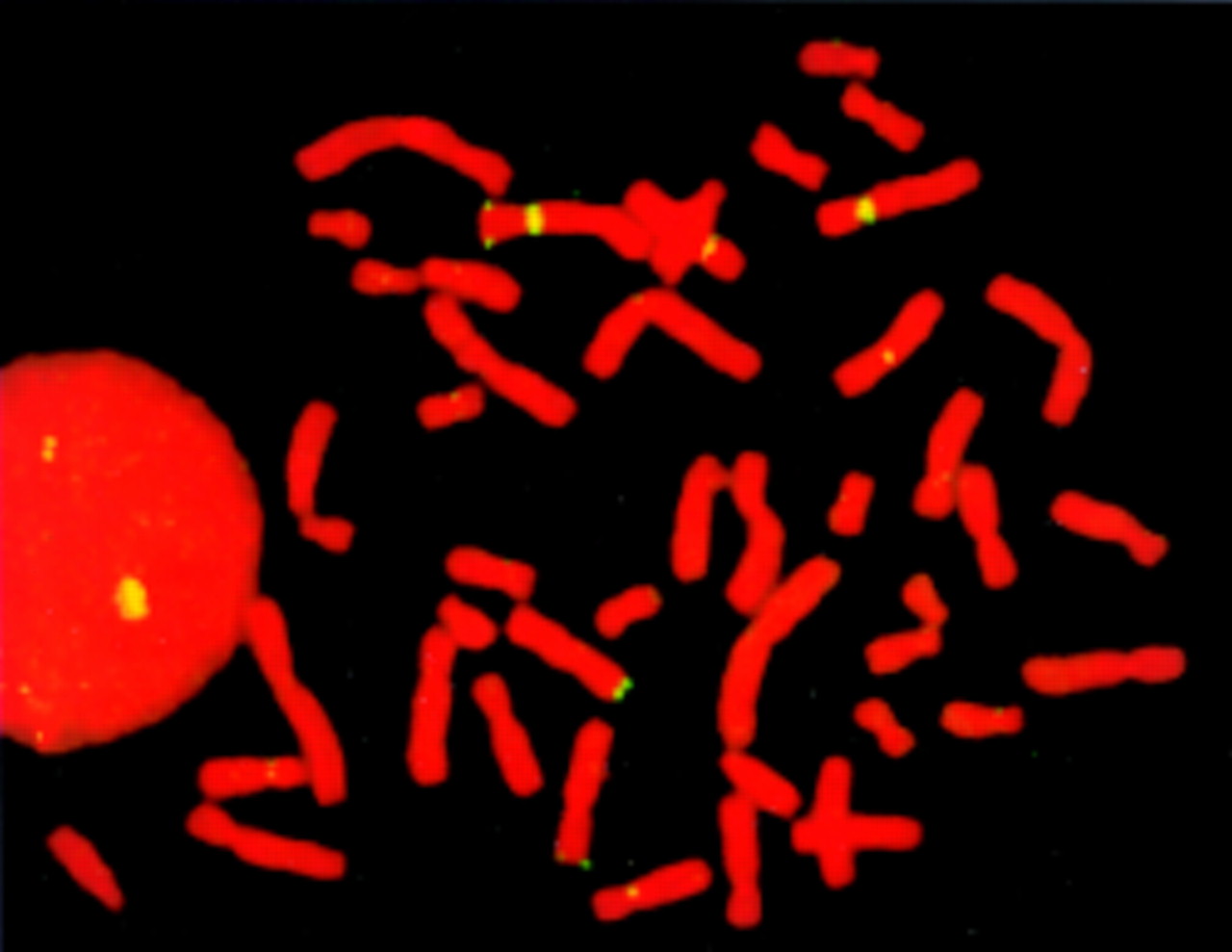
To identify the chromosome involved in the translocation of patient 6, we used different paints and telomeric probes of G negative bands. Additional signals on chromosome 4 were noted with distal 7p probe (Oncor) (fig 3). We determined the karyotype in patient 6 as 46,XY,der(4),t(4;7)(p16;p22).ish t(4;8)(wcp4−, 7ptel+, pC847.351−, D4S96−, 228a7−; wcp4−, 7ptel+). Balanced translocations in his parents were excluded with FISH using D4S96 and 7ptel in combination with pGXba11/340.

{kind=link}
{kind=link}
{kind=link}
FISH results in patient 6. FISH with distal 7p probe showing two signals in the distal region of 7p and an additional signal in the distal short arm of one chromosome 4.
The results of the analyses of polymorphic markers are listed in table2. Blood samples from the father of patient 5 were not available. Therefore, the size of the deletion was determined by FISH only. DNA from patient 4 was not available to identify the origin of the deletion.
In patients 1, 2, 3, and 6, the maternal allele was absent and in patients 4 and 5 the origin of the deletion remains undetermined. In patients 1, 2, and 6, the deletion 4p was at least 3.8 Mb in size and in patients 3-5 the size of the deletion can be estimated to be 13 Mb.
In our study of 22 patients with Wolf-Hirschhorn syndrome, a de novo translocation was present in 5/22 (22.7%) and a familial translocation in 3/22 patients (13.6%) (manuscript in preparation). In one additional patient (patient 5) a de novo translocation was suspected, but could not be confirmed. To our knowledge, such a high rate of proven de novo translocations in WHS has not previously been reported. This could be explained by the refined molecular-cytogenetic techniques such as FISH analysis and microsatellite analysis used here. In particular, de novo unbalanced translocations between the short arms of chromosomes 4 and 8 seem to be not uncommon in WHS. Müller-Naviaet al 10 described one fetus and Petit et al 11 12 another male patient with this chromosomal aberration.
The breakpoints of 4p and 8p differ in our patients, which makes sequence homologies between these chromosomal segments less likely. Thus, it is of great interest that Kogi et al 32 and Gondoet al 33 described a new class of tandemly repeated satellite DNA elements on chromosome 4p and 8p. They identified a tandem array of several RS447 sequence copies in 4p15 and distal 8p. This repeat was suggested to be responsible for formation of inversion duplication 8p34 and might probably explain the higher frequency of translocations between 4p and 8p.
Moreover, other chromosomes also, for example chromosome 7 in patient 6, occur in de novo unbalanced translocations involving the short arm of chromosome 4. Our findings support the hypothesis of Reidet al 35 that cryptic complex chromosomal rearrangements are more common than usually disclosed by light microscopy of conventionally stained chromosomes. Selection bias was unlikely, as we investigated blood specimens of all children with known WHS and with a tentative diagnosis of WHS.
Determination of the origin of the deletion showed that in all four patients in whom this was possible the maternal allele was absent, which is in contrast to published data. To avoid bias owing to familial translocations, which are mostly inherited maternally in a 2:1 ratio,36 the patients' parents were also investigated by conventional karyotyping and FISH analysis. We reviewed cases in which the origin of the 4p deletion was determined and found 27 paternally inherited and six maternally inherited deletions.7 15 37-45 Thus, 82% of reported patients with WHS show a paternally derived deletion.
As to the published patients with maternally inherited deletions, the patient Val described by Anvret et al 39 had a more complex chromosomal rearrangement causing WHS. Further cytogenetic investigations were not possible. Patients 3 and 6 from the report of Dallapiccola et al 44 and case 2 of Thies et al 42 presented with a de novo, maternally inherited deletion with breakpoints in 4p16.1, 4p16.3, or 4p13, respectively. However, FISH analysis with whole chromosome 4 paint was not performed to exclude a cryptic translocation. In the patient described by Greenberg et al,38 cryptic translocation in the mother was discussed, but not investigated. Patient 3 in the report of Kant et al 37 with maternal origin of the deletion had an unbalanced karyotype based on a cryptic translocation t(4;8) in the mother. Thies et al 42 described a third patient with de novo deletion and also a maternally inherited deletion. On the other hand, in two WHS patients with unidentified de novo translocations40 the deletion was of paternal origin.
Different explanations for the preponderance of paternally inherited deletions are possible, for example, statistical deviation or real biological phenomena such as increased mutation rates in sperm or genomic imprinting.41 An excess of rearrangements in male meiosis, related to differences between the mechanisms of sperm and egg production, have also been discussed.44
One aim of our study was to determine whether the WHS phenotype is influenced by the trisomic autosomal segment. The clinical findings in our patients 1-5, in previously published patients with familial translocations t(4;8),37 46-52 in patients with partial trisomy 8 owing to inversion duplication53-59 or direct duplication,60 61 respectively, and in patients with WHS6 62 are compared in table 3. There are several clinical signs which are usually present in patients with partial monosomy 4p as well as in those with partial trisomy 8p, such as mental retardation, muscular hypotonia, prominent forehead, broad nasal bridge, large mouth, dysplastic ears, and congenital heart defects. Thus, these features are by no means specific enough to distinguish between the phenotypes.
Clinical signs of patients with duplication 8p, with unbalanced translocation t(4p;8p), with monosomy 4p, with trisomy 7p, and reported patients
The face in WHS is much more characteristic than in partial trisomy 8p and is not significantly influenced by the presence of this trisomic segment. All in all, it seems to us impossible to differentiate clinically between patients with WHS resulting from monosomy 4p and WHS resulting from unbalanced translocation 4p;8p. This agrees with the conclusions of other authors.37 47-51
As to patient 6, to our knowledge, no other patient with a de novo translocation t(4p;7p) has been described before.63 64However, many patients with trisomy 7p resulting from familial translocations have been reported. These patients are not listed in table 3 because a considerable influence on the phenotype by monosomic autosomal segments cannot be excluded. Only those patients with distal duplication 7p are included.65-67 The characteristic, clinically recognisable phenotype in trisomy 7p encompasses delayed closure of fontanelles, sparse or even lack of eyebrows, a short nose with a low and broad nasal bridge, small upper and prominent, full lower lip, micrognathia, hypotonia, congenital heart defect, and delayed speech development.65 Macrocephaly and enlarged cerebral ventricles were only described in one patient.66One might speculate that relative macrocephaly in patient 6, which is highly unusual in patients with WHS, may be the result of the duplicated segment in 7p. Delicado et al 66 noted intestinal malrotation with dilatation of the sigmoid and left colon in their patient, but excluded Hirschsprung's disease. Our patient presented with similar anomalies of the intestines. We assume that this clinical sign, which has not been described in WHS before, may be caused by the duplication 7p. In conclusion, the overall phenotype in patient 6, in contrast to patients 1-5, appears to be considerably influenced by the trisomic region of 7p (table 3).
In summary, we show that de novo translocations causing WHS are more frequent than previously estimated. We recommend performing FISH analysis with wcp(4) in every WHS patient to exclude or confirm a de novo translocation as a common mechanism causing WHS. Also patients with cytogenetically visible deletions should be investigated by molecular-cytogenetic techniques to exclude cryptic rearrangements. Moreover, de novo translocations t(4p;8p) in WHS patients are more frequent than previously suspected.
Note added in proof
Since submission of the final version of this paper, 13 patients of this study have been published elsewhere.
Acknowledgments
We thank Professor Passarge for continous support and for reading the manuscript, and Professor Horsthemke for helpful discussion. In addition, we thank Barbara Henke, Barbara Ulrich, Elke Jürgens, and Gudrun Rodepeter for excellent technical assistance and the parents for taking part in this study. We thank Dr Herdit Schüler for the results of previous chromosomal analysis in patient 1, Dr A J H Hamers for performing FISH with D4S10 in patient 2, Dr J J M Engelen for FISH in patient 4, and Dr Schröder and Professor A Gal for SSCP analysis of the L1CAM gene in patient 6. We also thank Tracy Wright for the cosmids pC847.351, 33c6, 21f12, and 228a7. In addition, we thank CEPH for providing the YACs. DW and GG-K were supported in part by a young investigators grant from the Medical Faculty University of Essen (IFORES 107 402.0) and by the Deutsche Forschungsgemeinschaft (Wi1440/4-1).