Article Text

Statistics from Altmetric.com
- FRET, fluorescence resonance energy transfer
- HSCR, Hirschsprung’s disease
- LD, linkage disequilibrium
- SNP, single nucleotide polymorphism
The RET proto-oncogene, located at 10q11.2, encodes receptor tyrosine kinase expressed during neural crest development and is involved in different human neurocristopathies such as the multiple endocrine neoplasia type 2 (MEN 2) syndromes and Hirschsprung’s disease (HSCR).1 HSCR (OMIM 142623), or aganglionic megacolon, is a common developmental disorder characterised by the absence of enteric neurones in variable lengths of the distal gastrointestinal tract, resulting in functional intestinal obstruction. Although RET is considered to be the major gene involved in HSCR, only a subset of HSCR patients can be attributable to traditional germline RET mutations (50% of familial HSCR and 10–15% of sporadic cases in selected series,2,3 or 30% of familial and 3% of sporadic cases in a population based study4). In an even smaller subset of HSCR patients (5–10%), germline variants are present in other genes, generally related to the developmental programme of neural crest cells, such as the glial cell line derived neurotrophic factor (GDNF), neurturin (NTN), endothelin 3 (EDN3), endothelin receptor β (EDNRB), endothelin converting enzyme 1 (ECE1), transcriptional factors SOX10 and PHOX2B, and Smad interacting protein 1 (SIP1).5–7 In addition, RET modifying gene loci at 9q31, 3p21, 10q11, and 19q12 have been described associated with an HSCR phenotype.8,9 Because traditional germline mutations accounted for such a small subset of HSCR, we sought to determine whether there are more common susceptibility factors which predispose to a majority of HSCR cases. The silent variant A45A was described by our group in the only member from a MEN 2A/HSCR family co-segregating both phenotypes.10 When this variant in codon 45 (exon 2) and the RET haplotypes carrying it were found to be highly associated with a large subset of non-familial HSCR,11,12 it prompted us to propose that this was the associated variant itself, or that A45A was in linkage disequilibrium with the putative low penetrance susceptibility locus that would account for the majority of HSCR cases. Subsequently, we have shown that A45A anchors ancestral haplotypes in linkage disequilibrium, with the putative common founding susceptibility locus estimated to be 22 to 50 kb upstream.13 These observations have been confirmed in other populations,14–16 suggesting that this founder locus dates to the Stone Age.13
In this study we have undertaken systematic screening of the region upstream of the anchoring A45A single nucleotide polymorphism (SNP)—comprising RET intron 1, exon 1, and promoter—in 117 population based HSCR cases to find this putative founding locus.
METHODS
Patients and controls
In this study we included a series of 117 cases affected by clinically sporadic HSCR (20% female, 80% male), and their unaffected parents when available. More specifically, the triads (composed of the HSCR patient and both parents) were complete for 104 of the cases, while for 12 patients only one of the parents was available. There was only one patient for whom we could not collect DNA from either parent. The characteristics of our cohort of HSCR patients have been described in detail previously.11–13,17 In addition, we also included a group of 100 normal controls comprising unselected, unrelated, race, age, and sex matched individuals from Spain.
Key points
-
Several studies have shown that there are specific haplotypes of the RET proto-oncogene associated with the sporadic forms of Hirschsprung’s disease (HSCR). Based on such studies, linkage disequilibrium (LD) mapping estimates predicted the presence of an HSCR locus located at 22–50 kb upstream of codon 45 (exon 2) of RET. The aim of this study was to identify a founding locus responsible for the majority of HSCR cases in a particular series.
-
After a systematic mutational screening upstream of exon 2 of RET, the variants found were genotyped in 117 HSCR cases and 100 controls by fluorescence resonance energy transfer (FRET) or direct sequencing. Haplotype and genotype distributions were compared among these groups using standard case–control statistical analyses.
-
A specific RET haplotype was identified, clearly linked to HSCR phenotype. The LD of the analysed variants was maintained along the whole region studied up to position −1249. The ancestral haplotype associated with HSCR was characterised by the presence of specific single nucleotide polymorphisms (at −200 and −196) in proximity to the transcriptional start site. Functional modelling using luciferase expression assays showed a significantly depressed activity for the HSCR linked haplotype at −200/−196 in comparison with other combinations associated with controls.
-
These results seem to disprove the existence of an HSCR causing mutation as conceived in the traditional sense, but strengthen the concept of a specific combination of markers conferring susceptibility to the disease in a low penetrance fashion. It is conceivable that such an “HSCR haplotype”, together with other events occurring in other genes, might give rise to the disease. This would be in agreement with a polygenic model for the disease.
Informed consent was obtained from all the individuals studied, in accordance with the respective committees for protection of human subjects.
Identification, characterisation, and genotyping of RET sequence variants
Genomic DNA was extracted according to standard protocols.18 Mutational screening of the whole intron 1 and promoter of RET was undertaken by direct sequencing of overlapping fragments obtained by polymerase chain reaction (PCR), using an Applied Biosystems 3730 DNA sequencer, and aligning our results with the sequence provided in Genbank accession number NT 033985.
Large scale genotyping of each of the variants was done either by fluorescence resonance energy transfer (FRET) technology or by direct sequencing of the PCR products. The FRET internal probes were designed according to the sequences of interest and purchased to TIB Molbiol. Conditions for both FRET and sequencing, and the sequences of primers and probes, are available on request.
Statistical analysis
Allelic and genotypic frequencies of the RET polymorphisms analysed were calculated and then compared between patients and controls. Comparisons were made using χ2 analysis with Yates’ correction, with statistical significance set at p<0.05.
Haplotypes were constructed comprising 23 of the 42 novel variants (from −200A→C to IVS1+11637C→T) as well as the other three SNPs located in the 3′ end of the intron which had been analysed previously by our group.13 Generation of these extended haplotypes was possible and accurate because of the availability of the complete triad for 89% of the HSCR patients, which allowed us to reconstruct and compare the transmitted versus non-transmitted alleles. Based on the observation of complete linkage disequilibrium of an important part of the studied variants, and the existence of only a few combinations of these, it was plausible, and reliable, to infer the haplotypes for controls.
The haplotype distribution was compared between cases and controls. In addition, parental haplotypes were examined in the context of the affected children’s haplotypes, so that transmitted and non-transmitted haplotypes were noted and their frequencies compared.
Promoter constructs and assays
Plasmid construction for the RET minimal promoter region
The 5′ UTR region of the RET genomic DNA that comprises the variants −200A→G and −196C→A was PCR amplified using a forward primer (5′ GGGCTAGCCCGCACTGAGCTCCTACAC-3′) and a reverse primer (5′ CCCTCGAGCTGGAGGGACTGCGGCTA-3′). These primers span the minimum promoter region (−313 to −24 from translational start site) and both have a restriction site (NheI and XhoI respectively) at the 5′ end.
DNA samples with GA, GC, and AC (−200, −196) polymorphic variants were PCR amplified. The PCR products and the pGL3–basic luciferase report vector (Promega) were digested with NheI and XhoI. The fragments were cloned upstream of the XhoI site and transformed into E coli TOPO 10 competent cells following the manufacturer’s instruction (Invitrogen). Transformants with the desired fragment were screened both by restriction digest and by direct sequencing using Big-Dye Terminators v 20 cycle sequencing kit (Applied Biosystems), and analysed on an ABI Prism 3700 DNA analyser (Applied Biosystems).
Transient transfection and dual-luciferase assay
The dual-luciferase reporter assay system (Promega) was used to assess the activity of the RET promoter variants (indicated above). A human MTC cell line TT was seeded at approximately 6×105 cells in a six-well tissue culture plate with RPMI supplemented with 10% fetal calf serum. Cultures were incubated for 18–24 hours at 37°C in a CO2 humidified incubator.
When the cells were 40–60% confluent, they were washed and supplemented with serum-free media and transiently transfected and co-transfected with constructs and pRL-TK (Renilla plasmid as internal control). Transfection was carried out using DMRIE transfection agent following the manufacturer’s instructions (Life Technologies). Transfected cells were harvested 48 hours post-transfection and were lysed to carry out the dual-luciferase reporter assay according to the manufacturer’s instructions (Promega).
RESULTS
Novel SNPs and other types of polymorphic loci were sought upstream of IVS1–1463 by direct sequencing of the 23 kb intron 1, exon 1, and the region 12 kb upstream of the translational start site. This revealed 42 novel SNPs and two insertion/deletion polymorphisms (available on request and posted to the JMG website). Once identified, all the variants were tested in a small group of patients and controls and the preliminary results showed a strong association with HSCR. Because our fundamental aim was the identification of the putative functional low penetrance susceptibility locus for HSCR guided by our previous statistical calculation,13 we only genotyped the variants located nearest to the 5′ end of RET in the entire series of patients and controls. Following this strategy, we observed a lack of association of specific alleles with disease state at −1249C/T suggesting that linkage disequilibrium had broken (p = 0.08). Thus we subsequently concentrated on the region between −1249 and IVS1+11637, which was found to contain 22 novel SNPs and two insertion/deletion polymorphisms (table 1). We observed that the distribution of each variant was significantly different in HSCR cases from controls, showing an under-representation of all SNPs and an over-representation of the deletions (table 1, p<0.0001).
Allelic frequencies for the RET polymorphic loci in patients with Hirschsprung’s disease (HSCR) and controls
Because 89% of parental pairs were available for the HSCR cases, haplotypes comprising the analysed variants and those at the 3′ end of the intron (IVS1–1463T→C, IVS1–1370C→T and IVS1–126G→T)13 could be constructed for 92 of the cases and inferred for the rest. Although we found 13 different haplotypes among the HSCR cases and their parents, some of these were very rare (table 2). In fact, each of the 13 extended haplotypes could be defined as different subtypes within the five fundamental intronic haplotypes previously published (0–4).13 Of note, haplotype 0 (previously defined as “HSCR haplotype”)13 is always linked to a unique combination of alleles (table 2), and represents the most common haplotype among our cases (62%) compared with controls (17%).
Definition of the RET extended haplotypes
As parents of control subjects were not available, the haplotype construction for this group was inferred from the haplotypes of the HSCR cases, as described elsewhere.12,13 Using this method, haplotypes could be inferred for 82 of the 100 controls analysed. As shown in fig 1A, the difference in haplotype distribution observed in the cohort of patients and in controls was highly significant (χ2 = 76.96, p<0.00000001). In addition, we also calculated the haplotype frequencies among the non-transmitted alleles of the HSCR triads, and again observed significant differences when comparing transmitted alleles with HSCR patients v non-transmitted alleles (χ2 = 66.47, p<0.00000001; fig 1A). Conversely, the frequencies for each haplotype and the distribution are similar in the non-transmitted and the control alleles (χ2 = 0.88, p = 0.93).

(A) Distribution of the extended haplotypes in transmitted and non-transmitted alleles within the Hirschsprung’s disease (HSCR) families and in controls. (B) Distribution of the pairs of haplotypes in HSCR and controls.
We also compared the distribution of the pairs of haplotypes (genotypes) between patients and controls (fig 1B). Genotype 00 (42%) predominated in HSCR cases, while in the control group the most common genotype was 1a2a (21%), followed by 1a1a (15%). Perhaps the most interesting finding was that the “HSCR associated” genotype 00 was only present in 2% of controls.
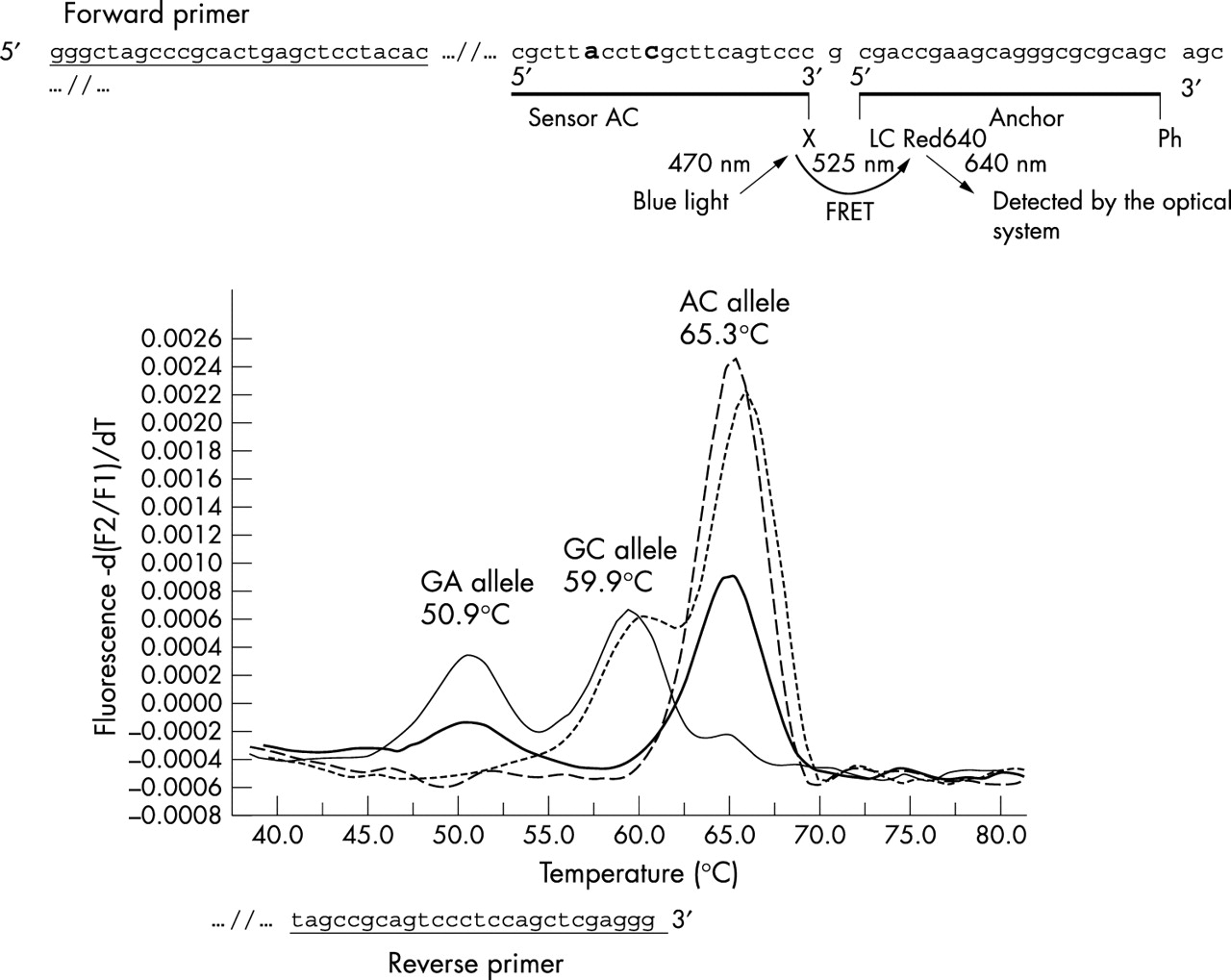
Because only two variants, −200A→G and −196C→A, lie within the promoter region and are in proximity to the transcriptional start site (at −195), we calculated the frequencies of the short promoter specific haplotypes comprising only the loci in this region in cases and controls. Such RET promoter haplotypes could easily be assigned for all the individuals tested because of the genotyping technique employed (see fig 4). As shown in fig 2A, the short promoter haplotype distribution was significantly different in cases and controls, as was the frequency of particular transmitted alleles to HSCR patients v non-transmitted alleles (χ2 = 83.64, p<0.00000001), or v control alleles (χ2 = 84.02, p<0.00000001). It is important to note that haplotype AC is in complete linkage disequilibrium with all the risk haplotypes described so far11–13 (table 2). In contrast, the frequencies for haplotypes GC (−200G −196C) and GA (−200G −196A) are considerably lower among HSCR chromosomes (25% and 15%) than among non-transmitted (44% and 40%) or control chromosomes (39% and 44%). Similar conclusions can be drawn from the comparison of the distribution of pairs of haplotypes (genotypes), where the differences observed are even more pronounced (fig 2B).
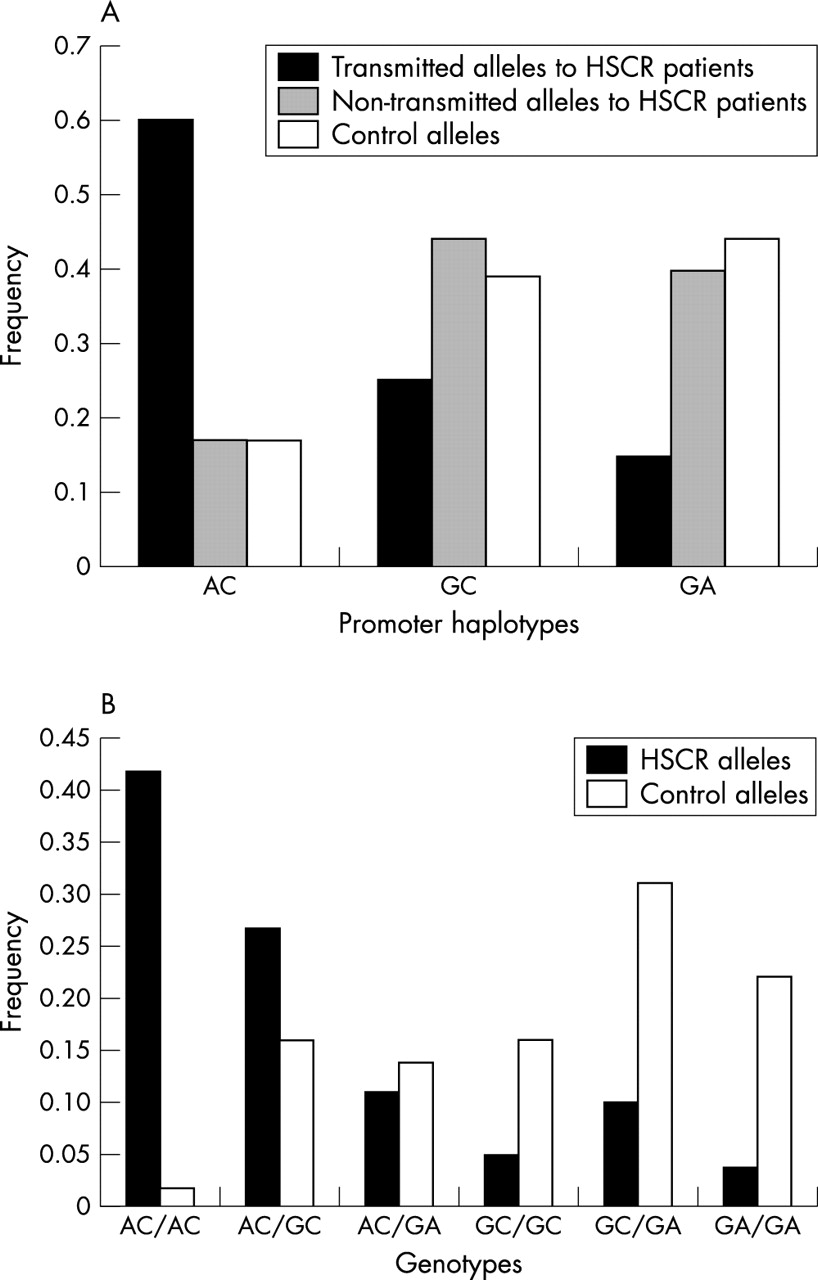
(A) Distribution of haplotypes comprising the RET promoter variants in transmitted and non-transmitted alleles within the Hirschsprung’s disease (HSCR) families, and in controls. (B) Distribution of pairs of haplotypes comprising the RET promoter variants in HSCR patients and controls.
Luciferase reporter assay of RET promoter function
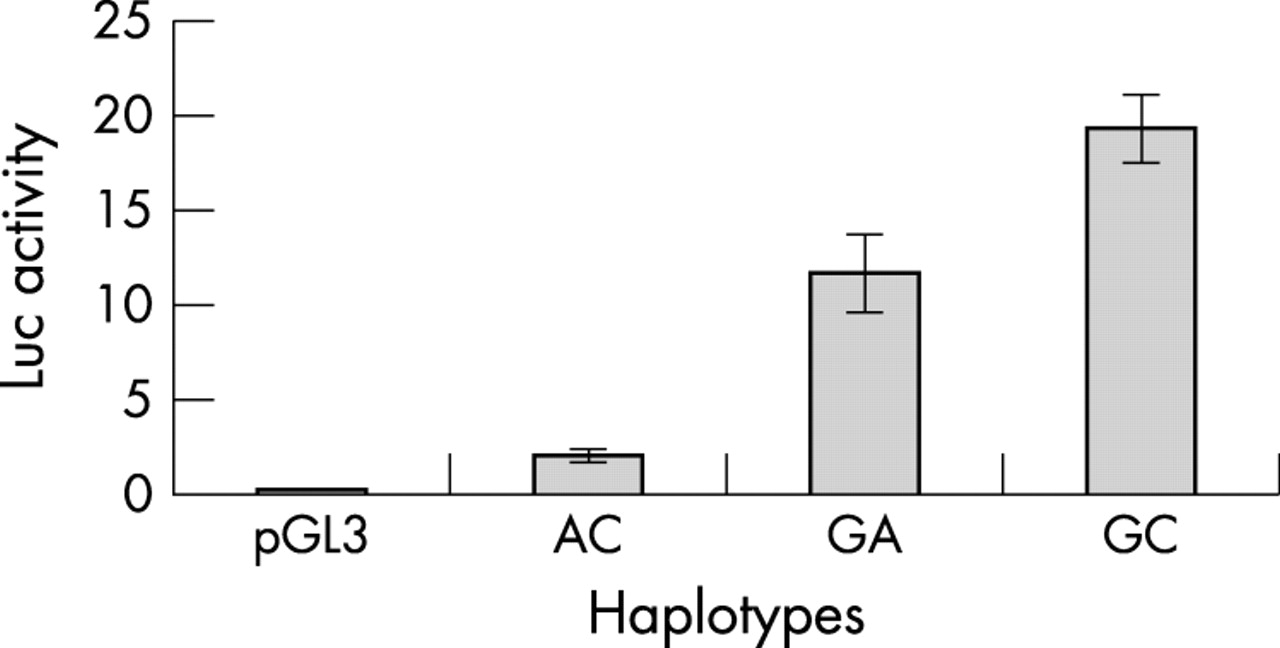
Luciferase activity was assayed in TT cells with constructs containing the three naturally existing promoter haplotypes at −200 and −196, GA, GC, and AC (see table 2). GC (or + − in table 2) had the highest expression (fig 3). AC (or − −), which is over-represented in HSCR, had the lowest expression (fig 4). Interestingly, GA (+ +) had an expression level less than that of GC but much more than AC, bringing its expression level much closer to that of GC than that of AC (fig 3).
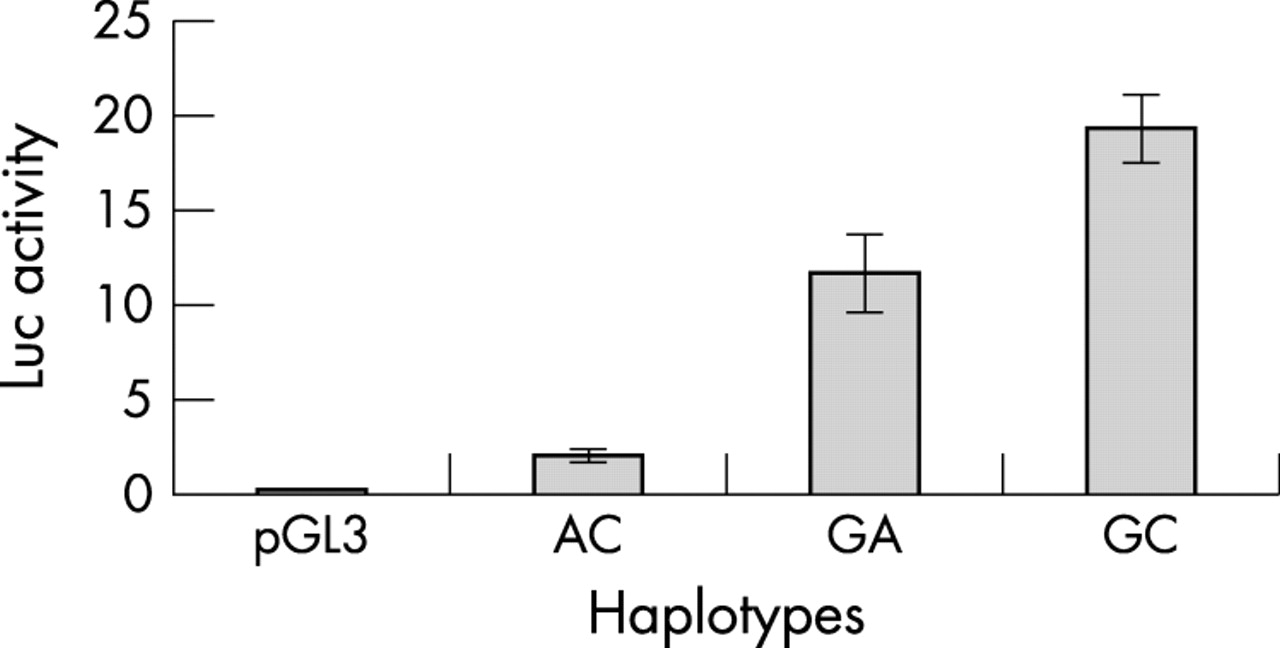
Luciferase assay showing decreased expression from the Hirschsprung’s disease (HSCR) associated −200 −196 haplotype AC (corresponding to − − in table 2) compared with the control associated GA (+ +) haplotype.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fluorescence resonance energy transfer (FRET) pattern for the simultaneous detection of the RET variants −200A→G and −196C→A.
DISCUSSION
It is currently well established that the RET proto-oncogene is the major gene implicated in the pathogenesis of Hirschsprung’s disease. Besides traditional germline loss of function mutations causing HSCR, several common RET proto-oncogene polymorphisms, present with different frequencies in different populations, have been associated with a variable risk of developing HSCR. In this sense, the HSCR associated variant firstly described was the silent polymorphism A45A, for which an association with the disease has been demonstrated in several cohorts of patients with different ethnic backgrounds, such as the Spanish, German, Italian, or Chinese.11,12,19–21
Several hypotheses have been put forward to explain the underlying cause of such an association, including a direct action of the variant or the existence of linkage disequilibrium with some functional, still unknown allele. While no functional assays have yet confirmed the first hypothesis, the second has gained major relevance because of the results of subsequent studies. More specifically, we observed that the c.135A allele, irrespective of the 3′ haplotype, was in linkage disequilibrium with a group of markers within the 3′ end of the first intron (haplotype 0: IVS1–1463T, IVS1–1370C, IVS1–126G).13 These results strongly suggested the existence of a low penetrance locus of susceptibility for HSCR located 5′ upstream all these markers.
Recently, the analysis of the promoter region of the RET proto-oncogene in three different populations of HSCR patients has revealed the existence of two new SNPs located at −5 and −1 from the transcription start site (−200A→G and −196C→A) in linkage disequilibrium with A45A.14–16 Moreover, there is a strong association with HSCR in such populations, supporting the postulate previously mentioned. In particular, the “ACA” haplotype was shown to be associated with HSCR and to present a significantly lower activity in dual-luciferase expression assays in vitro, compared with those haplotypes identified in the majority of normal controls.14 In agreement with these data, it has been suggested that the “ACA” combination may represent a core haplotype associated with HSCR, acting as a modifying risk allele in the development of the condition.14
Based on our previous results, our group has carried out a systematic mutational screening along the 5′ region of RET comprising the whole intron 1, exon 1, and the region 12 kb upstream of the translational start site, in order to identify the putative founding locus accounting for the majority of sporadic HSCR cases.13 Although no germline mutations were identified in our patients, we found several novel polymorphisms that are in linkage disequilibrium with the so called haplotype 013 and A45A, and strongly associated with HSCR. This association was supported not only by the results from comparative studies between HSCR cases and normal controls, but also from the TDT analysis in the HSCR triads. The linkage disequilibrium (LD) was maintained along the entire analysed region until position −1249, where the lack of association of specific alleles with the disease suggested that LD is broken. The relevance of our results resides in the finding of a putative LD breakpoint which would allow us to delimit the 5′ end of the “HSCR linked” RET region at a distance of approximately 24 kb with respect to A45A, in agreement with what it had been previously calculated by LD mapping estimates.13 This finding would be in contrast to the results of a previous study, in which the presence of a variant linked to A45A was described, located at approximately −4 kb from the translational start site.16 It would be plausible if the differences obtained in both studies reflected the distinct genetic background of the two populations examined. Future analyses of the region in different populations may help to elucidate this.
In conclusion, our findings seem to rule out the existence of an HSCR causing mutation, as conceived in the traditional sense, but strengthen the view that there is a specific combination of markers conferring susceptibility to the disease in a low penetrance fashion. The data derived from our functional in vitro studies, in agreement with other previously reported results,14 suggest that the haplotype 0 (table 2) may result in a lower level of expression of the RET gene. Thus homozygous individuals for this haplotype (62% of our cases and only 2% of controls) would present with considerably lower RET levels than those with other haplotype pairs. As the pathogenesis of HSCR is generally ascribed to loss of function or haploinsufficiency of the RET gene, the decrease in RET transcription rate associated with this haplotype could have a functional effect linked to RET messenger RNA expression. It is conceivable that such a “HSCR haplotype”, together with other events occurring in other genes, might give rise to the disease, which would be concordance with a polygenic model of the disease. However, the precise mechanism of actuation of this RET haplotype is not completely clear. Thus further studies are warranted to elucidate the functional relevance of this haplotype in the pathogenesis of Hirschsprung’s disease.
Acknowledgments
This study was partially funded by grants 1R01HD39058 from the National Institutes of Health, Bethesda, Maryland, USA (to GA, SB, and CE); Red de Centros 03/05 from the Instituto de Salud Carlos III, Spain (to RMF, GA, and SB); and CAA 56/03 from the Consejería de Salud de la Junta de Andalucía, Spain (to RMF, AP, GA, and SB). RMF is a doctoral fellow of the Fundación Ramon Areces. AP is supported by Instituto de Salud Carlos III contract CP03/00045. CE is a recipient of the Doris Duke distinguished clinical scientist award.
REFERENCES
Footnotes
-
Competing interests: none declared
Linked Articles
- Miscellaneous