Article Text

Statistics from Altmetric.com
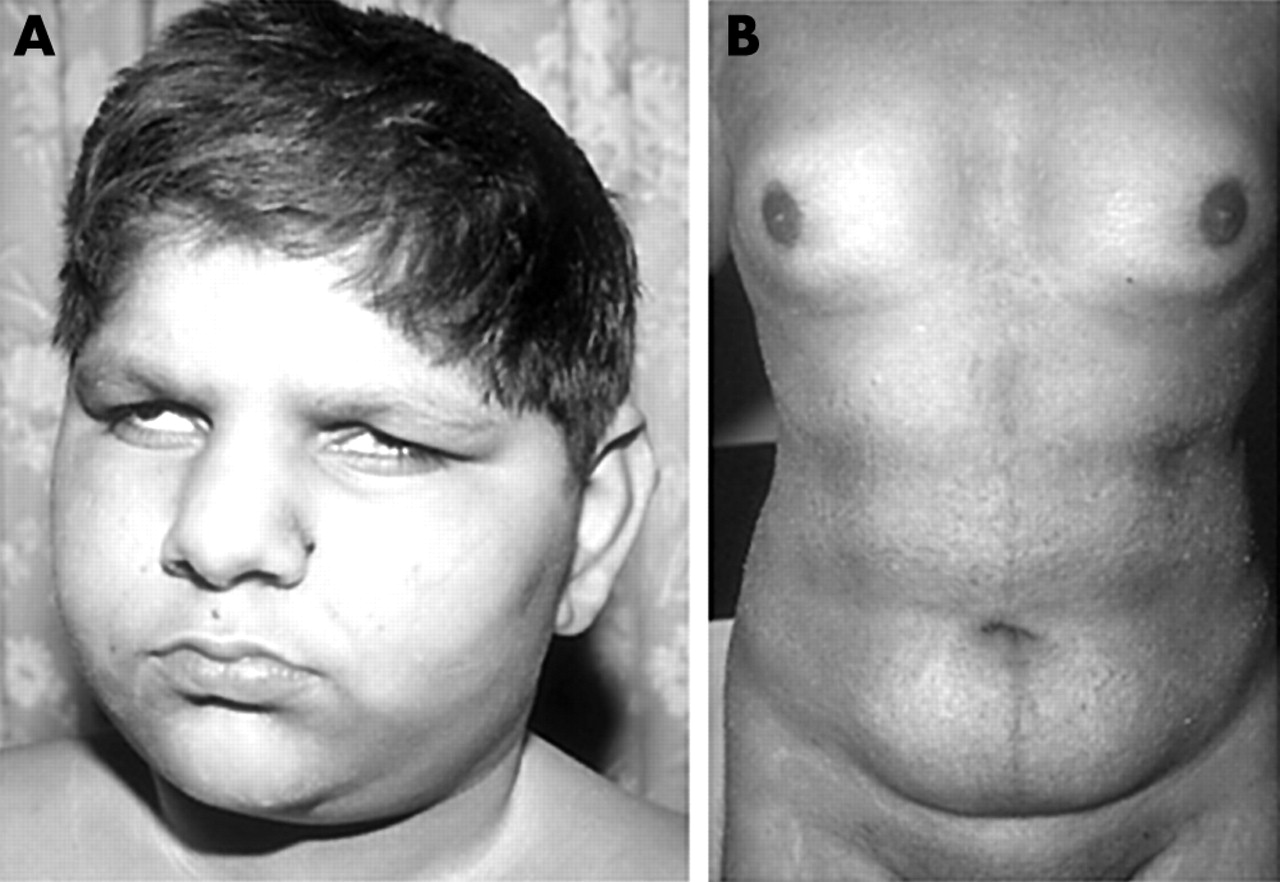
As part of a clinical study of Alström syndrome (MIM 203800) we sequentially ascertained seven families. Four of the families, pedigrees A–D (table 1), were consanguineous. In total there were 16 living affected individuals, aged 3–25 years. All had cone rod dystrophy that presented in the first 3 months of life with photophobia and nystagmus. The cone rod dystrophy progressed and all were registered blind by the end of the first decade. By the middle of the first decade a characteristic appearance of sunken eyes and a prominent supra-orbital ridge had developed (fig 1A). Truncal obesity became apparent in the first few years of life and all exhibited acanthosis nigricans in their teenage years. None has yet developed symptomatic diabetes. All males of sufficient age failed to enter puberty without hormone support and thereafter developed a female fat distribution (fig 1B). Deafness developed in all cases by the end of the first decade, but varied in severity and symmetry within and between families. All affected individuals were of normal intelligence though they experienced educational difficulties because of their combined and progressive sensory deficits. In all seven families other diagnoses had been made prior to the final diagnosis of Alström syndrome, presumably because of the rarity of the condition and the sequential presentation of disease features.1,2
Mutations in ALMS1 causing premature protein truncation in Alström syndrome patients exhibiting early onset cardiomyopathy
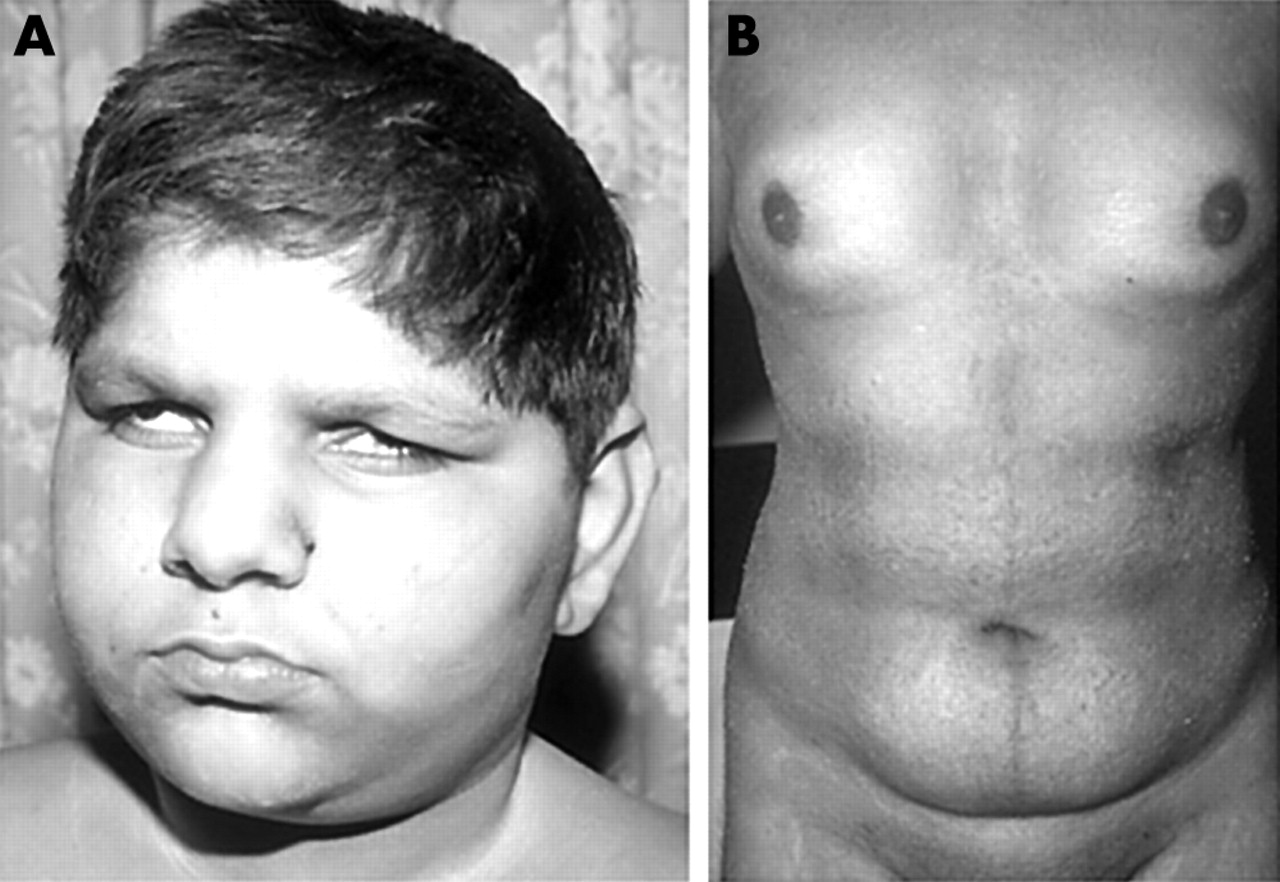
Face and body habitus of a study patient with Alström syndrome. (A) The face showing the deep set eyes, a feature that develops in the first few years. (B) The body of a teenage affected male, showing obesity and a female pattern of fat distribution. (Photographs reproduced with permission.)
In all seven families the index case had presented with symptoms of heart failure due to a dilated cardiomyopathy in the first year of life, usually within the first 3 months. One infant had been diagnosed as affected with Alström syndrome by prenatal diagnosis and was born with heart failure secondary to a dilated cardiomyopathy. All cases responded to conventional supportive therapy and the cardiomyopathy appeared to spontaneously resolve within 6 months. Although Warren et al first described dilated cardiomyopathy in Alström syndrome in 1987, the initial descriptions of the clinical features of Alström syndrome did not suggest that it was a frequent occurrence or that it could present in the first year of life.3,4 However, a more recent study found that 18 of 22 retrospectively diagnosed individuals had a dilated infantile cardiomyopathy.1 Taken together with our findings this suggests that infantile dilated cardiomyopathy may be a common feature of Alström syndrome.
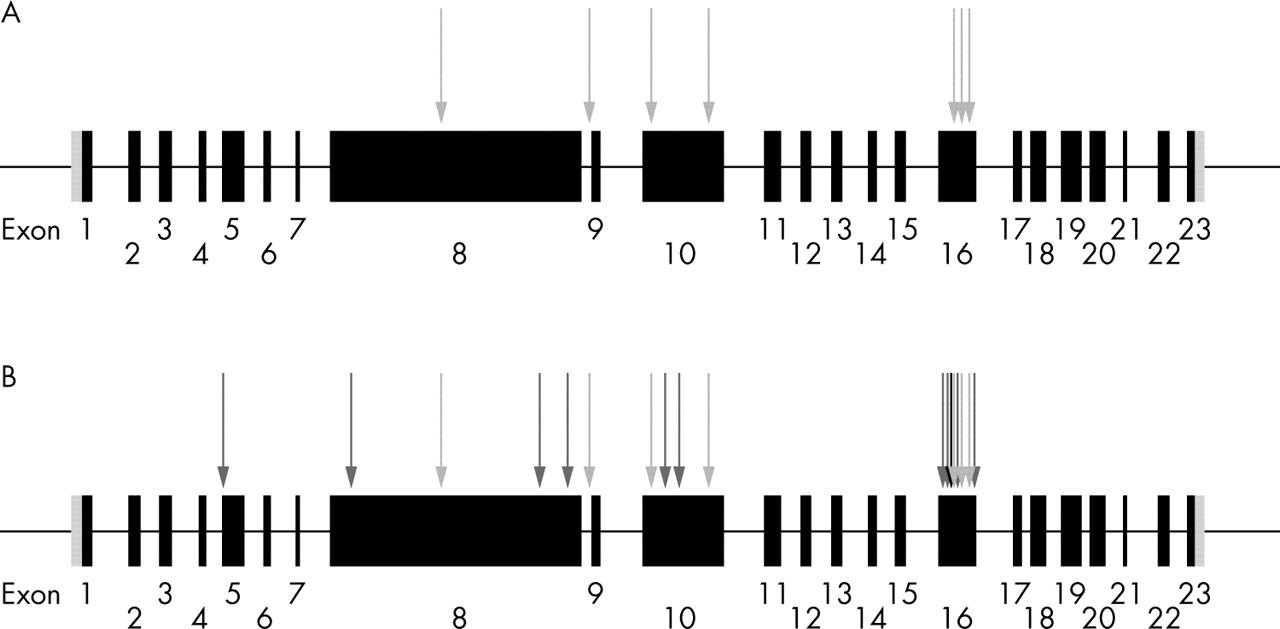
Alström syndrome appears to be monogenic, with all families reported linking to a single locus on chromosome 2p13.1.2,5–7 The 23 exon ALMS1 gene (GenBank NM_015120.2) spans 224 kb of genomic DNA and encodes a 4169 amino acid protein.5,6 All 14 previously reported ALMS1 mutations are predicted to cause premature protein truncation (fig 2).5,6,8 A single one of these mutations was stated to be associated with cardiomyopathy. We sought ALMS1 mutations in our seven families and compared the results to published mutations in which cardiomyopathy appeared less commonly associated.
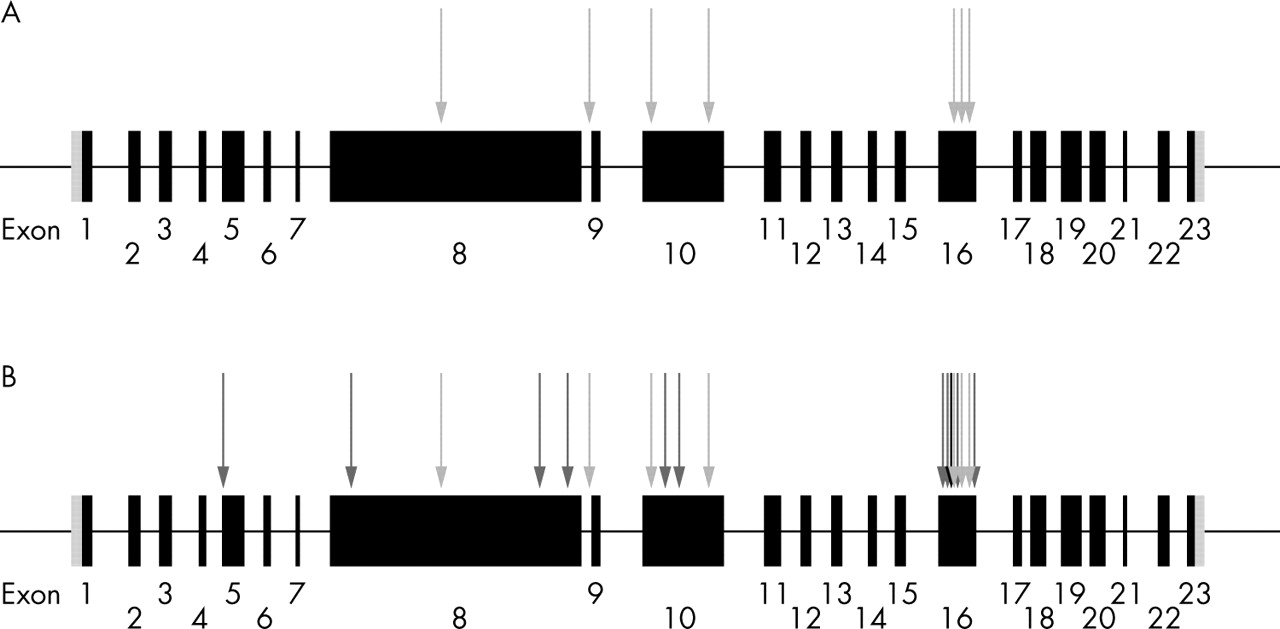
Schematic depiction of ALMS1 gene structure and distribution of mutations identified in Alström syndrome. Exons are shown as black filled boxes and untranslated regions as grey shaded boxes. (A) Novel mutations we report, associated with the occurrence of early onset cardiomyopathy (light grey arrows). (B) Distribution of all mutations identified in Alström syndrome (black arrow represents the position of a previously reported mutation associated with cardiomyopathy, dark grey arrows depict previously reported mutations which have no history of an association with cardiomyopathy, and our mutations are shown as light grey arrows).
Key points
-
We report seven families in which a diagnosis of Alström syndrome was eventually made and an ALMS1 mutation found. In each family the index case presented with a dilated cardiomyopathy usually in the first 3 months of life and prior to the appearance of other clinical features of Alström syndrome.
-
All ALMS1 mutations identified were predicted to cause protein truncation, including a homozygous exon deletion. There was no correlation between the occurrence of cardiomyopathy and the position of mutations within the ALMS1 gene.
-
We suggest consideration should be given to ALMS1 genotyping in all cases of dilated cardiomyopathy presenting in the first year of life. As over half of all reported ALMS1 mutations are located in exon 16, its analysis may serve as a useful initial screen for Alström syndrome.
METHODS
DNA was extracted using standard techniques from affected individuals and where available their parents. The local research ethics committee approved the study. All families had previously been genotyped using polymorphic micro-satellite markers flanking the ALMS1 gene and shown to be linked to the ALMS1 locus (data not shown). The gene was amplified and analysed in 44 fragments. Mutation analysis was primarily performed on parental genomic DNA using a Transgenomic (Omaha, NE) WAVE 3500A DHPLC machine (high pressure liquid chromatography) following the manufacturer’s methodology. DNA fragments showing alternate signature bands were sequenced using an ABI 3700 (Applied Biosystems, Foster City, CA) capillary sequencer. Exons 15, 20, and 21 were directly sequenced as they could not be optimised for DHPLC.
RESULTS
Pathogenic ALMS1 mutations were identified in all families (table 1; fig 2) and segregated as expected within the respective families (fig 3). In total seven novel mutations were identified in our cohort which were predicted to give rise to truncated protein products; five of these were nonsense mutations and two were deletions (table 1).

{kind=link}
{kind=link}
{kind=link}
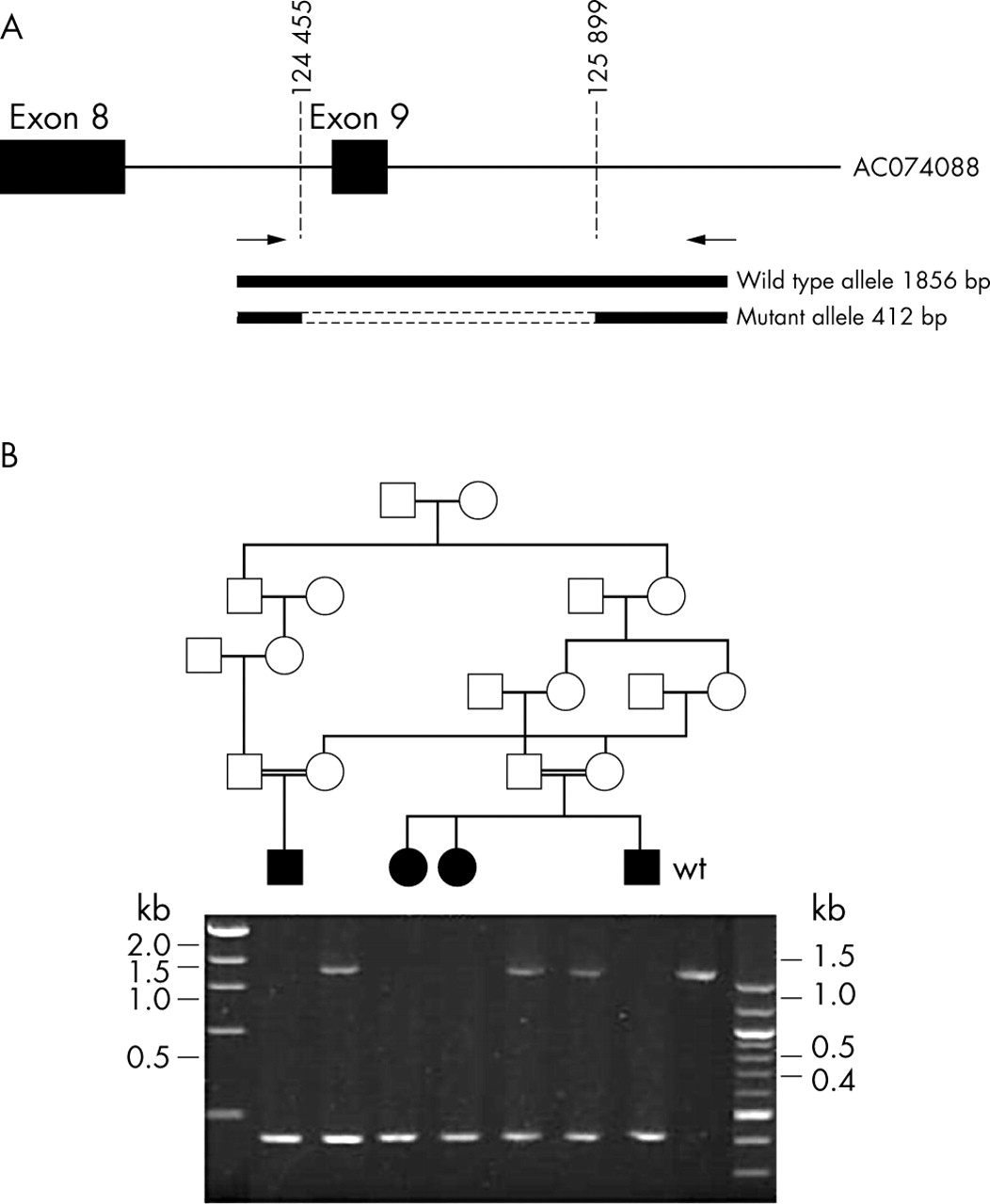
Schematic description of the IVS8+895del1444 mutation and its segregation within pedigree B. (A) Fragment of genomic DNA amplified to screen for the deletion. Forward and reverse primers shown as arrows and deletion breakpoints as dotted lines. (B) PCR products for the region containing the IVS8+895del1444 mutation. Parents (obligate carriers) showing the normal allele and the mutant allele (1856 and 412 bp, respectively) and the four affected children are homozygous for the mutated allele. Wildtype control is homozygous for the normal allele.
Initial studies of pedigree B suggested a deletion of exon 9. Further investigation showed that the affected children of this family had a homozygous 1444 bp deletion which encompassed the 134 nucleotides of exon 9 (IVS8+895del1444; intronic breakpoints corresponding to nucleotides 124455 and 125899 of BAC AC074008; fig 3). This deletion is predicted to create a new splice acceptor site (increase from 0.49 to 0.83 using the BDGP Splice Site Prediction by Neural Network programme available at http://www.fruitfly.org/seq_tools/splice.html) resulting in the introduction of 38 amino acids before a termination codon is created.
DISCUSSION
Comparison of the distribution of our novel cardiomyopathy associated mutations and those previously reported showed no obvious difference, with exon 16 being a mutational hotspot but the remaining mutations being spread throughout the gene (fig 2). Exon 16 (1163 bp) of the ALMS1 gene contains 52% (11/21 mutations) of all mutations described in Alström syndrome. There appears to be no genotype/phenotype correlation between mutation and the presence of cardiomyopathy in Alström syndrome. Analysis of the ALMS1 exon 16 mutations should be a useful initial screen for diagnosing Alström syndrome.
Dilated cardiomyopathy presenting in the first year of life has a heterogeneous aetiology and the outcome can be poor, but complete regression, as we report in Alström syndrome, often occurs.9,10 Our findings, and those of others, suggest that dilated cardiomyopathy is a common clinical finding in Alström syndrome. The cardiomyopathy may present symptomatically in the neonate. Alström syndrome should be considered in the differential diagnoses in a child presenting with infantile cardiomyopathy. The incidence of dilated cardiomyopathy presenting in the first year of life is approximately 80 per million children.11 Our experience of Alström syndrome suggests an incidence of 1.5 per million children, therefore we estimate 1–2% of cases of dilated cardiomyopathy presenting in the first year of life will proceed to a diagnosis of Alström syndrome. Making an early diagnosis of Alström syndrome gives important prognostic information to the cardiologist, defines the cause of the child’s illness, indicates that further disease features will develop, and informs the family of the one in four recurrence risk.
Acknowledgments
We would like to thank all the families who participated with this study and Alstrom Syndrome UK.
REFERENCES
Footnotes
-
↵* These authors contributed equally to the paper.
-
JB, JH, SS, ER, and CGW are funded by the Wellcome Trust.
-
Conflict of interest: none declared.
-
Ethical approval: the study was approved by the Ethical Committee of the Combined Leeds Health Care Trusts. Informed consent was obtained from all subjects involved in the study and when under 18 years of age, also from their parents.
Primer sequences used for sequencing ALMS1 and for confirming the exon 9 deletion are available from the corresponding author.