Article Text

Statistics from Altmetric.com
Rett syndrome (RTT; MIM 312750) is a neurodevelopmental disorder with an onset in early childhood that affects 1/10 000–1/15 000 females. After a period of relatively normal development, girls with RTT present with developmental arrest, usually by 12–18 months of age, followed by rapid deterioration with regression of speech and purposeful hand movements. Girls loose acquired communication skills coincident with the appearance of autistic features, deceleration of head growth, seizures, ataxia, awake respiratory dysfunction, and stereotypic hand movements.1
RTT is caused by mutations in the gene that encodes methyl-CpG-binding protein 2 (MECP2; MIM 300005).2 The involvement of MeCP2 in methylation specific transcriptional repression suggests that the symptoms of RTT are the consequence of inappropriate transcription of genes important for neuronal function and brain development. PCR amplification followed by direct sequencing of MECP2 coding exons 2 through 4 has become the standard diagnostic approach for clinically suspected RTT, but mutations are detected in only ∼85% of individuals with classic RTT.3,4 Gene dosage analysis by Southern hybridisation or multiplex ligation dependent probe amplification has resulted in identification of large genomic deletions of the MECP2 genomic locus in an additional subset of these mutation negative RTT patients.5–7 However, the causative mutation is still unknown in the remaining 5–10%.
It has recently been discovered that MECP2 has two splice variants encoding two protein isoforms that differ in their N terminus.8,9 The previously known mRNA splice variant (NM_004992) encodes a less abundant protein that is 12 amino acids shorter and translated from an ATG initiation codon in exon 2. The newly recognised transcript (AY541280) excludes exon 2 and encodes a longer protein translated from an ATG in exon 1, which was previously thought to be a non-coding exon.8,9 According to recently agreed nomenclature (Rett Syndrome Research Foundation annual meeting, June 28–30, 2004), these alternative isoforms have been renamed MeCP2_e2 (encoded by exons 2, 3, and 4) and MeCP2_e1 (encoded by exons 1, 3, and 4). MeCP2_e1 is much more abundant, especially in brain where its levels are 10 times higher compared to those of the MeCP2_e2 isoform. Prior to this discovery, mutation analysis of MECP2 was mostly limited to the coding sequences of exons 2, 3, and 4, but interestingly, MECP2 mutations in RTT in exon 2 were never found; they only affect exons 3 and 4 of the gene, thereby disrupting both isoforms. This is consistent with a more important functional role for the MeCP2_e1 isoform and supports the suggestion that exon 1 should be analysed in “mutation negative” RTT patients. To date, mutations in this exon have indeed been reported in three such individuals,8 but their overall prevalence in classic or atypical RTT remains undetermined. We therefore PCR amplified and sequenced exon 1 and flanking intronic sequences from DNA of 63 RTT patients with no previously identified mutations in the coding sequence of MECP2 exons 2, 3, and 4.
Key points
-
At least 85% of individuals with Rett syndrome (RTT) have mutations or deletions affecting the coding sequence of exons 3 or 4 of MECP2, but the mutations in the remaining individuals are still unknown. A recently discovered MeCP2 isoform excludes exon 2 but includes exon 1. The prevalence of mutations in MECP2 exon 1 in individuals with RTT is not well known.
-
We sequenced exon 1 from genomic DNA of 63 affected individuals (38 with classic RTT, and 25 with atypical RTT) who previously tested negative for mutations involving exons 2, 3, and 4. These individuals with classic RTT belong to an original cohort of 321 affected females. We studied X chromosome inactivation (XCI) using the androgen receptor assay on peripheral blood leukocyte DNA of patients with an identified mutation.
-
We identified a de novo mutation at the intron 1 splice donor site (c.62+1delGT) in a classic RTT patient. XCI studies revealed 68%:32% skewing with preferential inactivation of the paternal X. A second patient with atypical RTT has an 11 bp deletion (c.47_57del), creating a frameshift and premature stop codon (p.Gly16GlufsX36) and has random XCI. Surprisingly, although exon 1 belongs to the major MeCP2 isoform, mutations of this exon account for only 1/321 (0.3%) of classic RTT and 2/63 (3.1%) of all “mutation negative” RTT in this study.
-
Mutations in MECP2 exon 1 thus appear to be rare. However, inclusion of exon 1 should be incorporated into a comprehensive DNA diagnostic testing strategy for RTT.
METHODS
Patient material
Thirty eight patients had classic RTT and 25 had atypical RTT. Blood samples were obtained from patients and their parents under a protocol approved by the Institutional Review Boards of Baylor College of Medicine and its affiliated hospitals and the University of Alabama at Birmingham. We prepared total genomic DNA from peripheral blood leukocytes or from lymphoblastoid cell lines using standard protocols.10 These patients had no mutations affecting the coding sequence of exons 2, 3, and 4 of MECP2.
Sequence analysis
To PCR amplify exon 1 we used primers X1F: 5′-ccatcacagcc aatgacg-3′ and X1R: 5′-agggggagggtagagaggag-3′ as previously described.8 PCR products were purified using a commercial PCR purification kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Purified products were bidirectionally sequenced by automated fluorescent dye-terminator sequencing on an ABI Prism 3730xl sequencer (Applied Biosystems, Foster City, CA). Patient sequence data from both orientations were compared to the published reference sequence (AF030876) using pairwise Blast comparison (http://www.ncbi.nlm.nih.gov/blast/bl2seq/bl2.html). Sequencing electropherograms from patients and controls were electronically subtracted using Mutation Surveyor (Softgenetics, State College, PA) followed by visual inspection and confirmation of variant sequences. All detected sequence variants were confirmed on at least two independently amplified samples.
X chromosome inactivation
To analyse X chromosome inactivation (XCI) patterns, we used a modified protocol based on the previously described androgen receptor (AR) assay.11 In brief, 1 μg of genomic DNA was digested overnight with the methylation sensitive restriction enzyme HpaII (Roche Molecular Biochemicals, Indianapolis, IN). PCR was used to amplify 100 ng of either digested or undigested DNA using 32P-labelled primer. The amplified PCR fragment contains a HpaII site, as well as the highly polymorphic trinucleotide repeat of the androgen-receptor gene. Since the HpaII sites on the inactive X chromosome are methylated, only this allele is amplified following genomic restriction enzyme digestion with HpaII. The resulting PCR products were separated on 6% denaturing polyacrylamide gels and autoradiographed on Hyperfilm (Amersham International, Buckinghamshire, UK). The gels were also exposed to Fuji phosphorimaging screen and the intensity of the bands was quantified using a Model Storm 860 phosphorimager with imageQuaNT software version 5.2 (Molecular Dynamics, Sunnyvale, CA). Because the smaller allele amplifies more efficiently, a correction factor was generated using the undigested samples so that both alleles were represented equally. The pattern of XCI was determined by dividing the density of the smaller allele by the sum of the small and the large (corrected) allele. Assays were performed in duplicate for each sample.
Reverse transcription (RT)-PCR
Whole blood RNA was extracted using the PAXgene Blood RNA Kit (Qiagen, Valencia, CA). In brief, 1–2 μg of total RNA was reverse transcribed with Oligo(dT) using a standard MMLV reverse transcriptase protocol (Superscript II, Invitrogen, Carlsbad, CA). RT-PCR primer sequences for HF and HR1 and conditions were as described.8 Amplified products were subcloned in a TOPO-TA vector (Invitrogen, Carlsbad, CA), checked for insert size and sequenced with M13 sequencing primers. The sequence of primer HR2 is 5′-gcggccacggcggtcccact-3′. Amplification with HF and HR2 was performed at an annealing temperature of 61°C to yield a 121 bp product.
RESULTS AND DISCUSSION
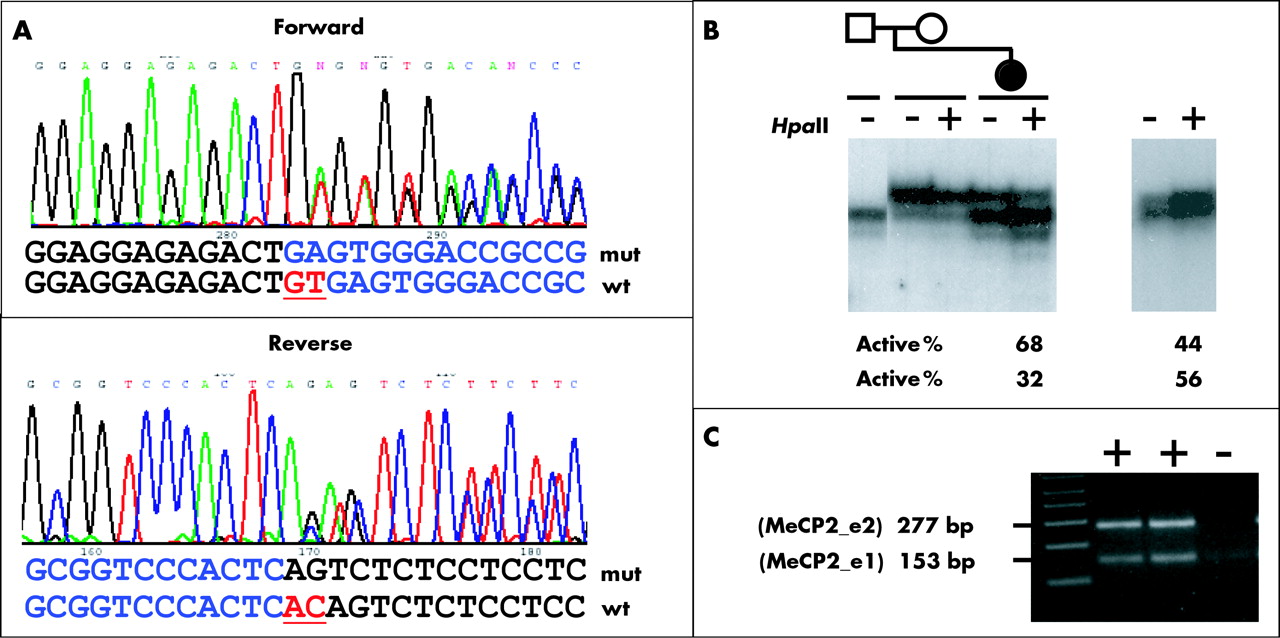
In the current study, we tested 63 RTT patients who had no mutation in MECP2 coding exons 2, 3, and 4 for mutations in exon 1 and flanking sequences. Thirty eight were from a total cohort of 321 classic RTT patients and 25 patients had atypical RTT. We identified a novel splice site mutation in an 8 year old female diagnosed with classic RTT. The mutation consists of a 2 bp deletion that removes the first two nucleotides (“GT”) of intron 1, thereby eliminating its consensus splice donor site (c.62+1delGT) (fig 1A). Analysis of parental DNA revealed that it is a de novo mutation, not present in either parent. The patient presented with pronounced hand dyspraxia and hand stereotypies, loss of language skills, and a developmental disturbance with relatively severe mental retardation, but interestingly, she displays good eye contact and a normal gait, suggesting a somewhat milder phenotype. XCI studies on a leukocyte derived DNA sample from the patient demonstrated a borderline skewing with preferential inactivation of the paternal allele (68%:32%) (fig 1B). Unfortunately, the MECP2 sequence of this patient is uninformative for known linked polymorphisms in the gene. Therefore, we could not determine whether it is the paternal allele that carries the mutation, but previous studies point towards a paternal germline origin of most de novo MECP2 mutations.12 The absence of the exon 1 splice donor site is not only predicted to disrupt the exon 1 containing MeCP2 isoform (MeCP2_e1) but could also potentially affect the expression of the exon 2 containing product (MeCP2_e2) because sequence from the 5 kb intron 1 could now be included in its 5′UTR.

(A) Mutation of MECP2 intron 1 in classic RTT. Portions of the forward (top) and reverse (bottom) electropherograms representing the sequences of the superimposed wild type (wt) and mutant (mut) alleles are shown. Deleted nucleotides are underlined. (B) Analysis of the X chromosome inactivation (XCI) patterns. Left panel: a partial pedigree of the family of the patient with the splice donor mutation, indicating the members analysed by the AR assay, is shown above an autoradiogram of labelled PCR products. The right panel shows similar XCI analysis on the patient with the 11 bp deletion. DNA from each individual was used directly as a template (− lanes) or digested with HpaII (+ lanes) prior to PCR amplification. For both affected individuals, two alleles of the polymorphic CAG repeat in the AR gene are seen in the undigested sample. The HpaII digested DNA sample from the patient with the 11 bp deletion shows random XCI (44%:56%). In the patient with the intron 1 mutation, the allele originating from the paternally inherited X chromosome is overrepresented after HpaII digestion, indicating preferential inactivation of this X chromosome (68%:32%). Note that for the affected individual’s mother, only one allele is present indicating homozygosity for the polymorphic CAG repeat. (C) RT-PCR on PBL RNA from the affected individual using primers HF and HR1.8 Both the upper 277 bp fragment (represents the MeCP2_e2 transcript) and the lower 153 bp fragment (represents the MeCP2_e1 transcript) were present at equal intensity.
Analysis of the genomic sequence flanking exon 1/intron 1 boundary, using the splice prediction program SpliceView (http://l25.itba.mi.cnr.it/~webgene/wwwspliceview.html), predicted with a high probability (0.87) that the next best ectopic donor splice site is 51 nucleotides downstream of the deleted splice donor sequence. This is only slightly less than the score of 0.88 calculated for the authentic splice donor site of exon 1. To test these possibilities, we performed RT-PCR on leukocyte derived RNA from the patient (fig 1C). We found that the expected amplified fragments from the normal MECP2_e1 and MECP2_e2 transcripts were present at equal levels. No amplified fragments of a different size that could have resulted from aberrant splicing at cryptic exonic or intronic splice sites were present. To improve detection of rare abnormally spliced transcripts, we also shotgun subcloned the RT-PCR products. Analysis of 40 clones did not show differently sized inserts and sequencing of 20 clones revealed only the wild type sequence. It is possible that we could not detect aberrant splicing or a change in the expression level of both transcripts because of preferential inactivation of the X chromosome carrying the mutant allele. Alternatively, the aberrantly spliced transcript could be highly unstable, for example due to nonsense mediated mRNA decay. We then performed RT-PCR amplification with primers HF and HR2 (fig 2), which should only yield a product if the predicted mutant transcript is present. Only one out of four replicate reverse transcribed cDNAs from the patient yielded a very low abundance amplification product (fig 2), which we were unable to successfully sequence. Based on tissue specific variation of XCI patterns13,14 and on previous studies suggesting random XCI patterns in RTT brain,15,16 the mutant allele might still be expressed at significant levels in brain. Conversely, a more skewed pattern of XCI in the brain could explain the patient’s slightly milder phenotype.

{kind=link}
{kind=link}
RT-PCR with new reverse primer HR2 on the patient with the c.62+1delGT mutation. HF and HR2 amplify the putative mutant transcript if the second 51 bp downstream predicted splice donor (*) was used. ptRT lanes show PCR amplification on four replicate reverse transcription reactions (+ lanes) and two without reverse transcriptase enzyme (− lanes) on leukocyte derived RNA from the patient. cRT lanes are similar reactions on a control RNA. gDNA lanes contain positive control amplifications from genomic DNA, H2O lanes contain no template, and unlabeled lanes are empty. Only a faint product is clearly seen in one of the ptRT (+) lanes (arrowhead).
In one atypical RTT patient we identified an 11 bp deletion (c.47_57del) which leads to a frameshift and premature stop codon (p.Gly16GlufsX36). XCI studies on a leukocyte derived DNA sample from this patient revealed a random pattern (44%:56%) (fig 1B). This mutation is identical to one recently reported in a classic RTT patient and is in a region with an AGG repeat, which could be prone to recurrent de novo deletions.8
The majority of RTT patients have mutations in exon 3 and 4 of the MECP2 gene, which affect both protein isoforms, while the mutations in exon 1 are specific for MeCP2_e1 only. To date, only three other cases with exon 1 mutations have been reported,8 but in none of them has the XCI pattern been described. It is interesting that in contrast to these unique exon 1 mutations, no mutations in exon 2 are currently known. A favoured explanation is that exon 2 mutations which exclusively affect the MeCP2_e2 isoform may not result in an RTT phenotype due to compensation by the high abundance MeCP2_e1 isoform. Conversely, disruption of MeCP2_e1 would be sufficient to cause a classic or atypical RTT phenotype.
Exon 1 mutations are rare in our series of patients. We found them in 0.3% (1/321) of all classic RTT patients and in 3.1% (2/63) of patients with all types of RTT, classic and atypical combined, who on prior testing did not have mutations in exons 2, 3, and 4. The overall low prevalence of exon 1 mutations which disrupt the N terminus and/or cause early truncations of the more abundant MeCP2_e1 isoform, suggests that they are more severe and possibly not compatible with survival unless some degree of XCI skewing is present. An alternative explanation is that exon 1 mutations are rarer in RTT compared to those in exons 3 and 4 because in some cases compensatory upregulation of the MeCP2_e2 transcript is present, preventing an RTT phenotype. This would be consistent with recent observations in mice where a null mutation of Mecp2 could be rescued by transgenic overexpression of the Mecp2_e2 cDNA.17 RT-PCR experiments on RNA from one classic RTT patient studied by us and one studied by Mnatzakanian et al,8 both with exon 1 mutations, did not show MeCP2_e2 upregulation. This may explain why the absence of only the MeCP2_e1 isoform led to RTT in these individuals.
Our data provide valuable information for mutation studies in RTT. Although mutations in exon 1 may be a rare cause of RTT, analysis of this exon should be included in mutation analysis of the MECP2 gene, in addition to sequencing of the other coding exons and a method to detect large genomic deletions.5–7 It still remains to be determined whether exon 1 is more commonly mutated in other conditions that can be associated with mutations in MECP2, such as autism and non-syndromic mental retardation.
REFERENCES
Footnotes
-
This work was supported by grants from the National Institutes of Health (NIH P01-HD40301) to IBV, HYZ, DGG, and AKP; the Tissue Culture Core of the Baylor Mental Retardation and Developmental Disabilities Research Center (NIH HD24064) to IBV and HYZ; by NIH HD 38985 to AKP; the International Rett Syndrome Association to DGG; and the Blue Bird Circle to DGG and AKP.
-
Conflict of interest: none declared.
-
Accession nos: Homo sapiens MECP2_e1: NM_004992, Homo sapiens MECP2_e2: (AY541280), Homo sapiens chromosome X MECP2 locus: AF030876.