Article Text

Statistics from Altmetric.com
- DAF, decay accelerating factor
- FH, factor H
- HUS, haemolytic uraemic syndrome
- MCP, membrane cofactor protein
- PCR, polymerase chain reaction
- RCA, regulator of complement activation
- TMA, thrombotic microangiopathy
Haemolytic uraemic syndrome (HUS), the most frequent cause of acute renal failure in childhood, is characterised by the association of acute renal failure, microangiopathic haemolytic anaemia, and thrombocytopenia.1,2 The majority of HUS cases occur after an episode of infectious diarrhoea, and are associated with Escherichia coli O157:H7 infection. However, atypical cases of HUS occur in the absence of infectious diarrhoea, although less commonly. Some are inherited in either an autosomal dominant or a recessive pattern and these patients often experience relapse and progress to hypertension and chronic renal disease. Sporadic forms can occur with many of the same signs and symptoms. The pathophysiology of this syndrome is poorly understood. However, after the first association with a low antigenic level of C3 was identified, two complement alternative pathway proteins, factor H (FH) and recently membrane cofactor protein (CD46; MCP) have been identified as fostering the development of atypical HUS.3–7 The human complement cascade can be activated through three different pathways: the classical pathway, the lectin pathway, and the alternative pathway. Factor H and MCP are regulatory proteins of the alternative pathway.8 Since the description of a genetic association of the disease with the cluster of complement related genes on chromosome 1, several mutations have been reported on the exons coding for the C-terminal domains of FH, which are important for C3b binding.5,9–12 Recently, two groups presented evidence that CD46 mutations may also predispose to HUS.7,13 Considering the major role of FH in complement alternative pathway regulation, we speculated that other complement regulators might be involved in the disease process. Human complement factor I (factor I) is a serine proteinase that cleaves the α chain of C3b and plays a key role in inhibition of the alternative pathway amplification loop which generates C3 convertase from C3b.14 We therefore studied the factor I gene in patients suffering from atypical HUS and having no factor H mutation.
METHODS
Patients
The subjects in this study were patients attending departments of paediatric and adult nephrology and departments of internal medicine in several university hospitals in France and Belgium over a period of 3 years. All participants provided informed consent. The clinical work out was performed in each referring centre. Twenty five patients (15 children and 10 adults) with atypical HUS and no factor H mutation were studied. Six of the patients had a familial variant of HUS (at least two siblings with the disease) and six exhibited persistent mild alternative pathway complement consumption with low C3 levels. Nineteen patients presented a sporadic, atypical form of HUS.
Key points
-
Atypical haemolytic uraemic syndrome (HUS) is a severe disease frequently leading to end stage renal failure. Complement factor H gene mutations have been reported in familial and sporadic HUS. However, some patients with atypical HUS show no evidence of factor H gene mutation, but instead exhibit alternative pathway-mediated complement consumption with decreased antigenic levels of C3.
-
We speculated that other complement regulators might be involved in the disease process and analysed the serine protease factor I which plays an important role in inhibiting the alternative pathway amplification loop.
-
We screened the complete coding sequence of the factor I gene, located on chromosome 4, in patients presenting with familial (n = 6) or sporadic (n = 19) atypical HUS. None had evidence of factor H mutation and six had mild alternative pathway complement consumption along with low C3 levels.
-
In two cases, a nonsense mutation was associated with heterozygous factor I deficiency as assessed by decreased factor I antigenic levels. In another case a heterozygous mutation was located at the serine site, a mutation that may lead to functional factor I deficiency.
-
This is the first demonstration that factor I is involved in genetic predisposition to the disease.
-
The role of complement in HUS remains a subject of debate. Our findings together with the involvement of factor H and CD46 strongly imply C3 cleavage regulation in the pathophysiology of HUS. Our findings further suggest that C3 cleavage regulation plays a key role in the disease process.
Case reports
Patient 1, a 32 year old Caucasian woman (P1), after pregnancy developed acute renal failure, hypertension, and haemolytic anaemia of rapid onset. Renal biopsy disclosed a pattern of thrombotic microangiopathy predominantly in the glomeruli, with few vascular lesions. HUS recurred 2 and 4 months later with partial renal function recovery but persistent hypertension. She was started on maintenance haemodialysis 1 year later.
Patient 2, a 17 month old Caucasian boy (P2), suffered from HUS with severe microangiopathic haemolytic anaemia, hypertension, and proteinuria. A relapse occurred 6 months later. At the time of writing, 2 years after onset, he requires anti-hypertensive treatment but his renal function is normal.
Patient 3, a 26 year old woman (P3), was hospitalised for recurrence of HUS following a second renal transplantation. She had already lost her first kidney transplant as a result of HUS recurrence coinciding with acute rejection. Renal biopsies confirmed histological features of thrombotic microangiopathy (TMA).
Complement assays
Laboratory investigations were centralised at the Department of Immunology, Hôpital Européen Georges Pompidou, Paris, a unit specialised in complement biology.
Freshly drawn EDTA plasma samples were obtained from all patients. EDTA plasma was prepared by centrifugation at 4°C and was kept frozen at −80°C until used. CH50 activity was measured according to standard procedures. Results were expressed as the percentage of mean values obtained with a reference plasma prepared from 100 healthy blood donors (normal range: 100±30%). Plasma concentrations of C4, C3, and factor B antigens were measured by nephelometry (Dade Behring, Paris La Defense, France). Normal values assessed with pooled plasma from 100 healthy donors were 220±120, 960±300, and 200±110 mg/l (mean±2 SD) for C4, C3, and factor B, respectively. Factor H antigen concentration was measured by a sensitive ELISA method as previously described.15
Factor I antigen concentration was measured by a sensitive ELISA method. Briefly, Nunc MaxiSorp ELISA plates (Nunc, Roskilde, Denmark) were coated with goat polyclonal IgG anti-human factor I (Calbiochem, Meudon, France). After washing and blocking free reactive sites with PBS containing 1% BSA, the plasma to be tested was added at a dilution of 1:1000 and incubated for 1 h at 37°C. After washing, the plates were then incubated with biotinylated goat anti-human factor I IgG prior to the addition of streptavidin-biotinylated horseradish peroxidase and further incubation for 30 min at 37°C. Enzymatic activity was revealed using the orthophenyldiamine substrate.
Results of factor I and FH antigens assays were expressed as the percentage of mean values obtained with the reference plasma (normal range: 100±30%).
Genomic factor I DNA sequencing
The DNA was extracted from whole blood using the proteinase K/phenol method. Uncloned genomic DNA was amplified by means of a polymerase chain reaction (PCR) using oligonucleotides flanking each exon. Primer sequences, length of the PCR amplified fragments, and temperatures of hybridisation used for each reaction are given in table 1. PCR products were purified using Multiscreen® plates according to the manufacturer’s procedure (Millipore, Molsheim, France). Direct DNA sequencing of the purified PCR products was then carried out using the Dye terminator cycle sequencing method (Applied Biosystems, Courtaboeuf, France) with a 96 capillary Sequencer 3700 (Applied Biosystems, Courtaboeuf, France). Sequence analyses were performed using Sequencher® software.
Factor I primer sequences, PCR annealing temperatures, magnesium concentrations, and sequencing conditions
Nucleotide and amino acid numbering was according to the published cDNA sequence (accession number M25615).16 Nucleotide no 1 is that located 29 nucleotides before the start of the peptide signal sequence. The start codon of the sequence after the peptide signal was used as the first amino acid of the protein.
RESULTS
Plasma complement profiles at the time of diagnosis are given in fig 1 and table 2. At the time of diagnosis patients 1 and 2 had low C3 and factor B levels. However, blood samples from patient 1, obtained at the time of end stage renal failure and from her parents, disclosed normal C3 levels. In contrast, patient 2 and his mother had permanently low C3 levels during the 3 years of follow up. Only one complement exploration could be performed in patient 3 after her second transplantation had been carried out and normal C3 levels were found. All patients had normal FH antigenic levels and no mutations were detected in 23 exons of the FH gene. Patients 1 and 3 and the father of patient 1 had decreased antigenic levels of factor I (50% of normal value), whilst patient 2 had normal factor I antigenic levels (table 2). Normal antigenic levels of factor I were found in 22 patients.
Complement studies of families 1 and 2 and patient 3
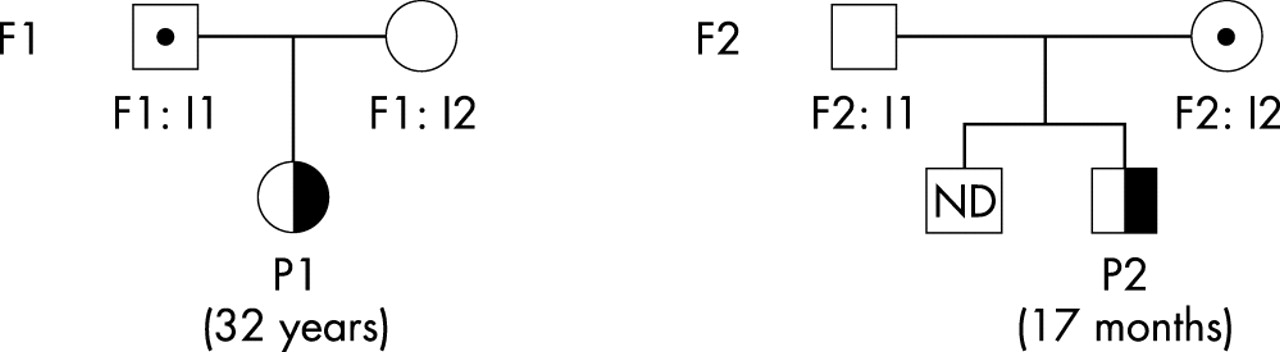
Pedigrees of patients 1 and 2. Families are indicated by F1 for patient 1 (P1) and F2 for patient 2 (P2). Age at onset is indicated in parentheses.
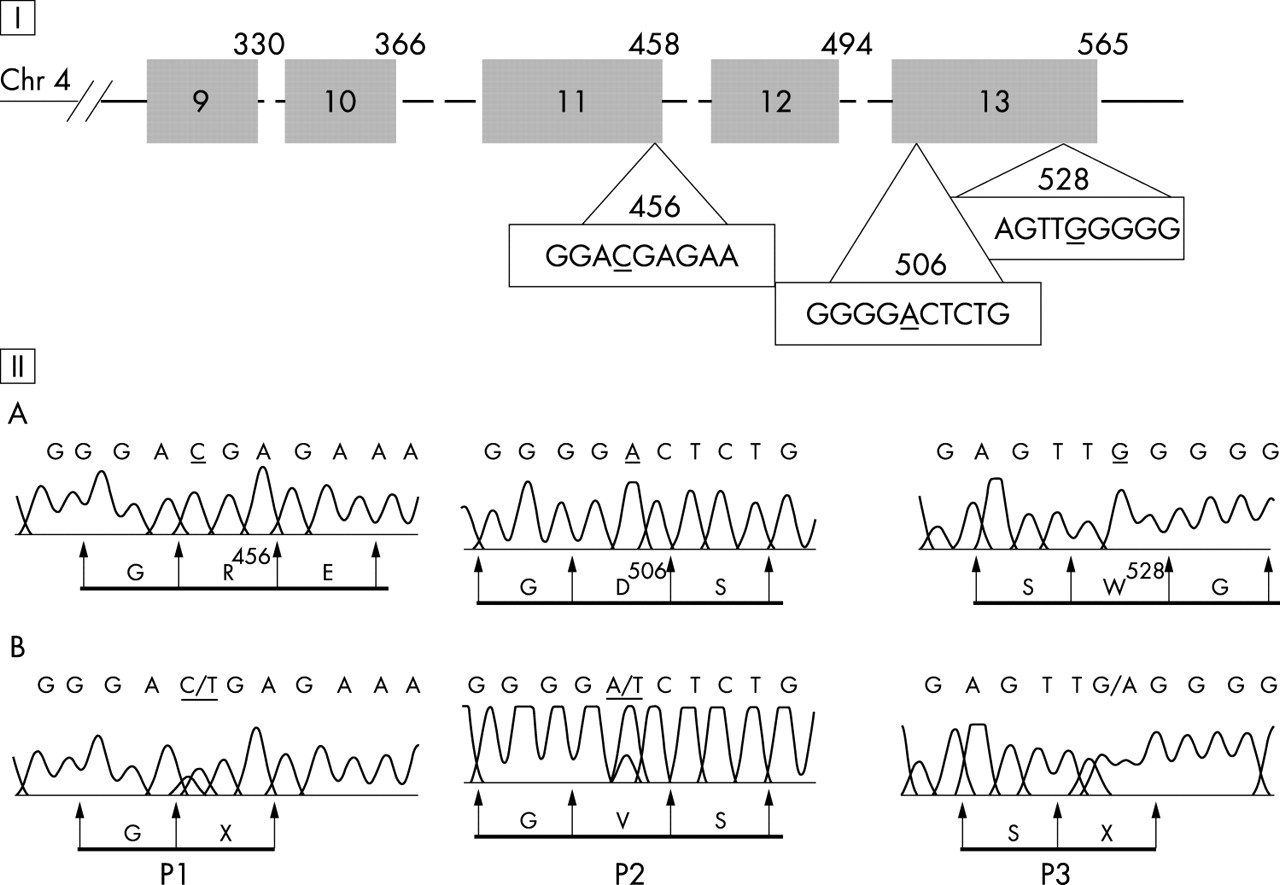
All 13 exons of factor I gene were amplified and directly sequenced using forward and reverse primers. Results of genetic analysis in the three patients are illustrated in fig 2. They were confirmed on two different samples, except for patient 3. Each mutation described was the only genetic abnormality found in each patient. In two cases, the molecular abnormalities found led to a nonsense mutation. In patient 1, we detected a heterozygous nonsense mutation at codon 456 caused by a nucleotide substitution from C (CGA for arginine) to T (TGA for stop codon) at nucleotide 1366 in the cDNA sequence. The healthy father of patient 1 was heterozygous for the same transversion. We identified a heterozygous G to A transversion of the second nucleotide of codon 528 in the genomic DNA of patient 3, leading to a stop codon (TAG) instead of a tryptophan (TGG). In family 2, a nucleotide substitution involving an aspartic acid (D) located close to the serine site was found in exon 13. Patient 2 and his mother presented a heterozygous nucleotide substitution (A/T) at codon 506 (GAC) leading to substitution of an aspartic acid for valine.
{kind=link}
{kind=link}
Genetic studies of factor I. (I) The light chain factor I gene is shown including the serine protease domain. The five exons are shown as boxes, and the introns are depicted by dotted lines (exons 9–13). The codon number according to the mRNA factor I sequence (accession number M25615 in GenBank) is indicated at the upper right hand corner of each box. (II) The positions of the mutations are shown. Wild type (A) and patient DNA sequences (B) (P1 for patient 1, P2 for patient 2, and P3 for patient 3) and corresponding amino acids are shown. P1: a heterozygote C1449T; this translation leads to a stop codon. P2: a heterozygote A1600T; this translation leads to an amino acid change D506V. P3: a heterozygote G1666A; this translation leads to a stop codon.
Factor I gene screening in the 100 healthy controls revealed the presence of four polymorphic variants in exons 11–13. One variant (1610C/A) has no consequence at the amino acid level (silent mutation; one heterozygous C/A). We identified three polymorphic variants A1246G (two heterozygous G/A) and G1351A (two heterozygous A/G) in exon 11 and C1671G in exon 13 (one heterozygous G/C) which cause an amino acid exchange (causing arg388hist, lys423arg, and glu530gln changes). The mutations located at the arginine 456, tryptophan 528, and aspartic acid 506 did not occur in the control group (200 chromosomes investigated). Consequently, these findings were interpreted as germline mutations.
DISCUSSION
The serine protease factor I cleaves the chains of C3b and is thereby involved in the regulation of the alternative complement pathways. The factor I gene is located on chromosome 4q25 and spans 63 kb. It comprises 13 exons and there is a strong correlation between the exonic organisation of the gene and the modular structure of the protein.17,18 We report for the first time a mutation in factor I gene affecting patients with atypical HUS. The patients’ mutations are located in exons 11 and 13 of the gene which codes for the light chain of factor I that contains the serine proteinase region of the molecule. We identified two heterozygous mutations, leading to a stop codon, which were associated with a decreased antigenic level of factor I. The abnormal gene encodes a putative truncated factor I protein that lacks the C-terminal amino acids including the serine proteinase region. These results demonstrate a heterozygous factor I deficiency. The serine proteinases are a large group of biologically important enzymes that have been extensively studied. This category of enzymes includes trypsin, other complement proteins, C2, C1r, C1s, factor D, and factor B, and proteins of the fibrinolytic and coagulation cascades.19 Factor I contains the catalytic triad of amino acids, aspartic acid, histidine, and serine, a triad common to all serine proteinases. Comparison of the serine proteinase amino acid sequences on the basis of crystallographic data and consensus secondary structure predictions showed that the aspartic acid 524 residue in factor I belongs to the five highly conserved amino acids GDSGG in the serine site. Mutation of the aspartic acid located at -1 of the active serine to valine may lead to functional factor I deficiency.
By its action on C3b, factor I impedes the formation of the alternative pathway C3 convertase (C3bBb). Thus, factor I has an important role in inhibiting the amplification loop of the alternative pathway which generates C3 convertase from C3b. The function of factor I depends on various cofactors. The cleavage of C3b depends on complement factor H, C4-binding protein, and cell-surface molecules such as complement receptor 1 (CD35) and membrane cofactor protein (MCP, CD46).20–23 In hereditary deficiency of factor I, the alternative pathway is not regulated, leading to a consumptive depletion of C3 and factor B. Complete hereditary deficiency of factor I has been reported in at least 30 different pedigrees and is associated with severe pyogenic infections. Only two different molecular lesions that elicit factor I deficiency have been previously identified.24 In one pedigree two siblings with factor I deficiency have the same missense mutation within the serine proteinase region of factor I. In the resulting protein leucine replaces histidine 400, a semi-conserved amino acid of the serine proteinase family. In the second pedigree, the proband was found to be a compound heterozygote. One allele carried the same mutation as identified in the first family, while the second allele had an unusual donor splice site mutation that caused skipping of the sequence encoded by the fifth exon from its processed transcript.
Two patients exhibited, at the time of diagnosis, evidence of alternative pathway-mediated complement consumption with low plasma levels of C3 and factor B. In one patient who was found to be factor I deficient, levels of C3 and factor B were within normal ranges. It is of note that in one patient the level of C3 was sometimes normal and thus masked partial factor I deficiency. As previously reported, levels of C3 and factor B may vary among FH-deficient individuals.15 Therefore, normal C3 and factor B levels do not rule out a diagnosis of heterozygous factor I deficiency. The mother of patient 2, who exhibited clear evidence of alternative pathway consumption, and the father of patient 1, both carrying mutations, have not yet developed clinical evidence of HUS. Incomplete penetrance is also a feature in HUS pedigrees with FH mutations and common polymorphisms of FH may contribute to HUS manifestation in subjects with and without FH mutations.25
A subgroup of patients with HUS has emerged with mutations identified in the complement factor H gene. In 1998, Warwicker et al undertook a candidate gene linkage study in two families with autosomal dominant HUS and in one family whose pedigree was compatible with recessive inheritance, or dominant inheritance with partial penetrance.5 The disease segregated with the region of chromosome 1q containing the candidate gene factor H in the regulator of complement activation (RCA) gene cluster. FH is the primary fluid phase regulator and inhibits the formation of the alternative C3-convertase and accelerates its decay. FH deficiencies have been reported in both homozygous and heterozygous forms.15 Genetic studies have also found several different heterozygous missense mutations between SCR16 and SCR20 suggesting a particular role of the C-terminal domains of the protein relevant to the pathophysiology of HUS.9,10,26,27 Neumann et al found FH mutations in 13% of 111 patients with atypical HUS.12 The RCA genomic region contains several candidate genes for HUS including the five human regulators of complement activation proteins: FH, complement receptor type 1 (CR1, CD35), membrane cofactor protein (MCP, CD46), decay accelerating factor (DAF), and C4b-binding protein.28 Recently, MCP mutations were detected by Richards et al in affected individuals of three families and by Noris et al in one family with low C3 levels.7,13 On the other hand, we can speculate that a link between the candidate proteins may be ascribed to the alternative pathway regulation. The alternative pathway is controlled by both proteins in the RCA family and by factor I whose gene is located outside the cluster. Furthermore, we observed persistent low C3 levels without evidence of mutation in all FH exons in patient with HUS. We conclude that a subset of patients with atypical HUS probably suffers from a genetically heterogeneous predisposition syndrome, resulting primarily from a dysregulation of the alternative pathway. So far, the functional consequences to renal cells are unknown.
The clinical presentation and outcome in our patients appear similar to those reported in patients with HUS and mutations in FH gene. The time of disease onset ranges from childhood to adulthood. The clinical course is complicated by relapses and the disease develops to end stage renal failure. Whilst factor H mutation is reported in cases of familial and sporadic HUS, the CD46 mutation is observed in familial HUS. In our patients a factor I mutation was only observed in cases of sporadic HUS. We did not find factor I mutation in six patients with a family history of the disease and we speculate that the penetrance of the disease may be low. In one of the three patients (patient 3), the kidney transplant failed twice following recurrence of the disease. Currently, renal transplantation in a patient with FH mutation apparently has a high risk of failure, whereas no recurrence of the disease was observed in patients with MCP mutation.7,29
CONCLUSION
Our data suggest that mutations in complement factor I predispose to atypical HUS. This is the first report of heterozygous factor I deficiency-associated disease. Three proteins, factor H, CD46, and factor I that participate in the regulation of the complement alternative pathway are now known to be involved in the genetic predisposition to atypical HUS.
Acknowledgments
Marine Arlie and Dominique Senger provided expert technical support. We are indebted to Professor Jurg Schifferli, Professor Michel D. Kazatchkine, and Professor Laurence Weiss for helpful discussion. We thank Professor S. Meri and Professor Alain Meyrier for reviewing the article.
REFERENCES
Footnotes
-
Conflict of interest: none declared.