Article Text

Statistics from Altmetric.com
First described by Maroteaux and Lamy in 1966,1 mucolipidosis III (ML III) or pseudo-Hurler polydystrophy (MIM 252600; Online Mendelian Inheritance in Man (OMIM), McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), 2000; http://www.ncbi.nlm.nih.gov/omim/) is a rare autosomal recessive condition resembling Hurler syndrome with no organomegaly or mucopolysacchariduria.
Clinical manifestations are variable and progress into adulthood. Bone involvement is slowly progressive and bone pain and disability due to destruction of hip joints are the most frequent and debilitating symptoms. Clinical symptoms include stiffness of the fingers and shoulders, claw hand deformity, short stature, and scoliosis. Mild coarsening of the face with corneal clouding, mild retinopathy, astigmatism, and cardiac valve involvement have also been reported. The radiological findings include moderate to severe dysostosis multiplex with vertebral changes.2
ML III is due to the abnormal trafficking and subcellular localisation of lysosomal enzymes.3–5 Newly synthesised lysosomal enzymes are secreted into the extracellular medium instead of being properly targeted to the lysosomes because they lack a mannose 6-phosphate receptor recognition marker.3,4 The UDP-N-acetylglucosamine-1-phosphotransferase enzyme activity is defective in ML III6 and three complementation groups (A, B, C) have been reported suggesting genetic heterogeneity.7 ML III complementation group C (MIM 252605) is characterised by deficient enzyme activity when assayed with lysosomal glycoproteins as acceptors, but normal activity when assayed with the simple acceptor, α-methylmannoside.6,8 In 1996, Bao et al9 showed that UDP-N-acetylglucosamine-1-phosphotransferase is a multimeric enzyme made up of three different subunits.9 We have previously ascribed ML III type C to mutations in the UDP-N-acetylglucosamine-1-phosphotransferase gamma subunit gene (GNPTAG) on chromosome 16p.10 Here, we report the genomic structure of this gene and its mutations in 14 additional patients belonging to eight families of various ethnic backgrounds. We suggest that, although the exact relative frequency of each ML III type is not known, the UDP-N-acetylglucosamine-1-phosphotransferase gamma subunit gene apparently plays a major role in mucolipidosis type III.
METHODS
Patients
The 14 patients reported here belong to eight families of Jewish Tunisian (3/8), Druze (1/8), Turkish (1/8), Dutch (1/8), Finnish (1/8), and Jewish-Iranian origin (1/8). Five families were consanguineous. The diagnosis of ML III was based on a combination of clinical, radiological, and biochemical criteria.2 The clinical criteria included: short stature, progressive joint contractures, mild mental retardation, fine corneal opacities, valvular heart disease, thoracic and spine deformities, and coarse facies.2
Key points
-
Our work aimed to identify new mutations in the UDP-N-acetylglucosamine-1-phosphotransferase gamma subunit gene (GNPTAG) and to collect clinical data on 14 patients affected with mucolipidosis type III.
-
We studied the 11 exons of the GNPTAG gene by direct sequence analysis in a group of 14 patients belonging to eight families of different background origin.
-
We observed six new mutations in the GNPTAG gene. Mutant genotypes included missense, frameshift, and splice mutations in Druze (1/8: G106S), Jewish-Tunisian (3/8: c.500insC), Jewish-Iranian (1/8: c.445delG), Turkish (1/8: c.608insC), Finnish (1/8: c.379del13bp), and Dutch patients (1/8: IVS5-1G>C and R66X).
-
We emphasise the aortic and mitral valve involvement in mucolipidosis III patients. A careful cardiac evaluation in order to detect severe valvular complications is recommended.
-
This study strongly suggests that the gamma subunit of the UDP-N-acetylglucosamine-1-phosphotransferase gene might play a major role in mucolipidosis type III.
Radiological inclusion criteria were dysostosis multiplex with unique pelvic and vertebral changes.2 Biochemical criteria consisted in an at least 10-fold increase in serum lysosomal enzyme activity level (arylsulfatase A, beta-hexosaminidase, alpha mannosidase) and/or low intracellular lysosomal enzyme activity level in cultured fibroblasts (14/14 patients).
Most patients presented with hand stiffness and claw hand deformity (11/14), stiffness of shoulders (7/14), scoliosis and/or kyphosis (8/14), and short stature (7/14), with pectus carinatum and/or thoracic asymmetry (6/14) (table 1). Hip pain was a major complaint in four patients (aged 17, 28, 33, and 35 years). Patient 5-II presented with hip osteochondritis while his asymptomatic brother (5-III) and sister (5-IV) were diagnosed on the basis of systematic biochemical tests performed because of the family history.
Clinical findings and mutations of the 14 patients studied
All the patients presented with variable degrees of dysostosis multiplex and progressive hip destruction. Low iliac wings with hypoplastic bodies, flattening and irregularity of the proximal femoral epiphyses, and underdevelopment of the anterior part of the vertebral bodies of the lumbar spine were also observed (11/14). Cardiac ultrasound showed a thickened aortic valve with aortic regurgitation in three of these patients. One patient (2-I) needed valve replacement therapy at the age of 27 years. A 17 year old patient (3-I) with severe aortic regurgitation is scheduled for surgery. Four patients had coarse facies, one had mild mental retardation (6-II), and one (7-I) had learning difficulties. Corneal opacity and lipoid degeneration were documented in only one patient (4-I) at the age of 25 years. We also noticed that four patients had a metallic voice.
Molecular studies
Genomic DNA was extracted from peripheral blood leukocytes of the affected individuals and parents by standard methods obtained after informed consent. PCR amplification of the 11 exons of the gene was performed in a total volume of 50 μl, containing 100 ng of template DNA, 1×PCR reaction buffer (Roche), 200 μM of each nucleotide, 75 ng of each primer (table 2), and 1.5 U of Taq DNA polymerase (Roche). DMSO 1% was added to all the reactions except for exons 1, 2, and 3 where DMSO 10% was used. Experimental conditions were: denaturation at 96°C for 2 min, then 35 cycles of denaturation at 95°C for 15 s, annealing at 60°C for 30 s, extension at 72°C for 30 min. In the final cycle, the extension lasted 10 min. PCR products (5 μl) were analysed by electrophoresis and visualised by ethidium bromide staining.
Exon–intron boundaries, sizes and PCR primer sets for amplifying the GNPTAG gene
Direct sequencing of amplification products was performed on an ABI PRISM 310 sequencer. The results were compared to control sequences and to the sequence of clone LA16-316G12. For web-based database homology searches, we used BLAST and FASTA software.
RESULTS
Clinical manifestations
The clinical findings and mutations of the patients studied here are presented in table 1. Increased serum lysosomal enzyme activity level and/or low activity level in cultured fibroblasts were present in 14/14 patients. Phosphotransferase activity towards artificial acceptor for phosphate (α-methylmannoside) was normal which was consistent with the diagnosis of ML III complementation group C in most of the patients. Unfortunately, we could not study the other Druze and Turkish patients.
Genomic organisation of the gamma subunit unit UDP-N-acetylglucosamine-1-phosphotransferase (GNPTAG) gene
The sequence of the gamma subunit of the UDP-N-acetylglucosamine-1-phosphotransferase gamma subunit gene (GNPTAG) has been reported in a previous study.10 A computerised BLAST study using the 5′ end of this cDNA sequence allowed us to identify a 100% similarity with the CAB56184 mRNA at locus AF302786 (EMBL: AL031709, AF302786, AE006467). Alignment of this mRNA with the genomic sequence of clone LA16-316G12 showed that the gene consists of 11 exons spanning 11.13 kb of genomic DNA. Interestingly, the average intron size is 200 bp, while the size of the third intron is 9 kb. This structure was consistent with the exon prediction of fgenes and Genscan. Each splice donor and acceptor site of the gene contained the canonical consensus sequences (table 2). Recently, the GNPTAG has been assigned to clone MGC 26283 and IMAGE 4824054 and reported as CAB56184, c316G12.3, or GNPTAG.11
Mutations analysis
Mutations on both alleles were detected by direct sequencing of the 11 exons of the GNPTAG gene in 14/14 patients. All the mutations segregated with the disease. They were located between exons 4 and 8 of the gene. The mutant genotypes are summarised in table 1. They include six novel mutations, namely, three frameshift mutations (c.608insC, c.445delG, c.379del13 bp), one nonsense mutation (R66X), one missense mutation (G106S) and one splice mutation (IVS5-1G>C). The patients were homozygotes for the mutant genotype in 7/8 families. Patient 8-I was a compound heterozygote for a paternally inherited splice mutation (IVS5-1G>C) and a maternally inherited nonsense mutation (R66X). RT-PCR experiments showed that the mutation (IVS5-1G>C) caused the skipping of exon 6 (fig 1). Patient 1-I was homozygous for the G106S missense mutation. His parents were heterozygotes and study of 100 chromosomes derived from healthy unrelated individuals from the same ethnic background failed to identify this G106S amino acid change. Interestingly, the previously reported c.500insC mutation,10 is described here in three families of Jewish-Tunisian origin.

Effect of IVS5-1G>C in patient 8-I. (A) Mutation IVS5-1G>C at the genomic level. (B) RNA was extracted from fibroblasts (RNA extraction kit, Qiagen). Reverse transcription (AMV reverse transcriptase, Boehringer) was carried out according to the manufacturer’s instructions using the following primers (RT–PCR): forward (c.254): GTTCCACAACGTGACCCAGCA and reverse (c.580): TGATCAGCTCATCGGCCAGGT. (C) Skipping of exon 6 at the c.DNA level in patient 8-I, compared to a normal control (D).
DISCUSSION
Here we report on GNPTAG mutations in 14 patients belonging to eight unrelated ML III families. We have previously reported on the c.500insC mutation in three families of Druze and Muslim origin. The cytosine insertion, reported here in Jewish-Tunisian patients belonging to three families occurred in a repeated sequence of cytosine and in a CpG island, which might have triggered the recurrence of this mutation. The c.608insC mutation (exon 8) also occurred in the context of a six-cytosine repeat; this could be due to slippage during DNA replication resulting in gains or losses of repeats. The two frameshift mutations, that is the c.445delG (exon 7) and c.379del13 bp (exon 6), led to a premature stop codon at the same 161 codon. The splice mutation (IVS5-1G>C) caused the skipping of exon 6 (fig 1). This mutation as well as the nonsense mutation (R66X) is predicted to severely damage the protein product.
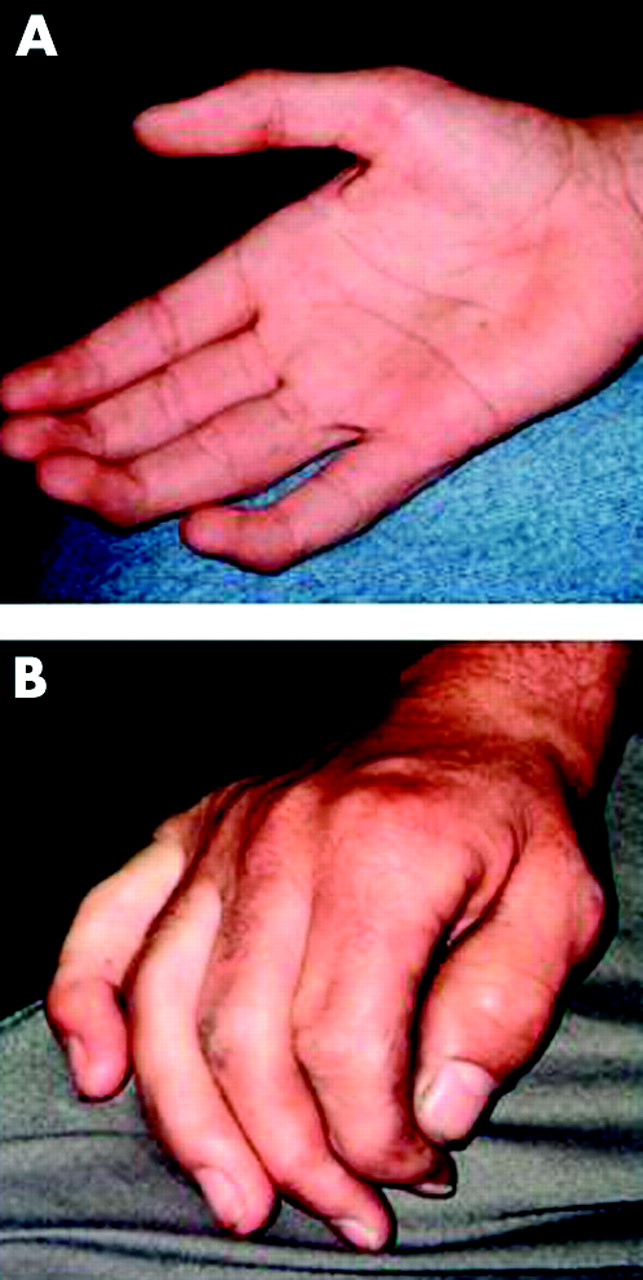
Here, we also emphasise joint and cardiac involvement in ML III. Stiffness of the small finger joints and/or claw hand (fig 2) was present in 11/14 patients. Hand involvement was already present in young patients (5-I, 7-I) at the age of 5 and 6 years and should be regarded therefore as suggestive of ML III. Freisinger et al12 reported on two ML III sibs with isolated involvement of the hip joint and no symptoms of storage disease. Here patient 5-II also presented with isolated hip involvement. On the other hand, three patients reported here had a thickened aortic valve and aortic regurgitation. The cardiac involvement in patients with ML III has already been reported by Kelly et al2 and Satoh et al.13 This suggests that ML III patients should undergo a careful cardiac evaluation and follow-up in order to prevent severe valvular complications.

{kind=link}
{kind=link}
(A) Claw hands of patient (3-I) at the age of 12 years and (B) hand of patient 2-II at the age of 35 years.
Acknowledgments
We thank Dr Yaacov Frishberg, for his help in the preparation of the manuscript.
REFERENCES
Footnotes
-
This study was supported by a grant from the Vaincre les Maladies Lysosomales Association (VML), France, and by a grant from the Keren Meshoutefet of the Hebrew School of Medicine, Jerusalem (ARR). Additional support was from the Crown Human Genome Center at the Weizmann Institute of Science (WIS) and an Israeli Ministry of Science Culture and Sports grant to the National Laboratory for Genome Infrastructure at the WIS (EBA).
-
Conflict of interest: none declared.