Article Text

Statistics from Altmetric.com
Neurofibromatosis 2 (NF2) is an autosomal dominant disease associated with mutations in the gene NF2.1 In clinical practice, the diagnosis is made if a patient satisfies the 1987 National Institutes of Health diagnostic criteria, which are either the presence of bilateral vestibular schwannomas or a first degree relative with NF2 and a unilateral vestibular schwannoma, or any two of the following: meningioma, schwannoma, glioma, neurofibroma, or juvenile posterior subcapsular lenticular opacity.2 These strict criteria have since been slightly relaxed to incorporate more patients who do not quite fulfil the diagnostic criteria but are thought nevertheless to have NF2.34
We report here a patient who was found to have NF2, by the strict criteria, and who had severe intellectual disability. Further investigation revealed a de novo balanced reciprocal translocation. We have fine mapped the chromosome 22 breakpoint and its relation to NF2 and established the effect of this translocation in various tissues from the patient.
METHODS
A 26 year old woman presented at hospital with acute abdominal pain. She was mentally handicapped since birth but was otherwise fit and well, living in a residential home with an excellent quality of life. Her speech and language were limited but she was able to communicate with those she knew well. At operation a large abdominal schwannoma was identified. She was subsequently found to have bilateral vestibular schwannomas on MRI and thus satisfied the diagnostic criteria for NF2.
GTL banding at the 550-band level was done using standard techniques. The X-inactivation status of the patient was determined by a polymerase chain reaction (PCR) based X-inactivation assay at the AR locus.56 DNA isolation from genomic clones and fluorescence in situ hybridisation (FISH) methods were used, as described by Cox et al.7 Clones from the RPCI-1, 5, 11, and 13 BAC and PAC libraries were obtained from BACPAC resources. Cosmid M0531 was identified from a chromosome 22 specific cosmid library prepared at the Lawrence Livermore Laboratory and was obtained from the German HGMP Resource Centre and Primary Database (RZPD). Exons 1–17 and the flanking intronic regions of NF2 were PCR amplified and sequenced on an ABI-377 machine using the ABI Prism BigDye™ kit, according to the manufacturer’s protocol (PE Applied Biosystems, Foster City, California, USA). Sequence was determined using primers designed in both directions, based on the published sequence, accession No Y18000. Southern blot analysis of the coding region was done using published primer pairs.8
RESULTS
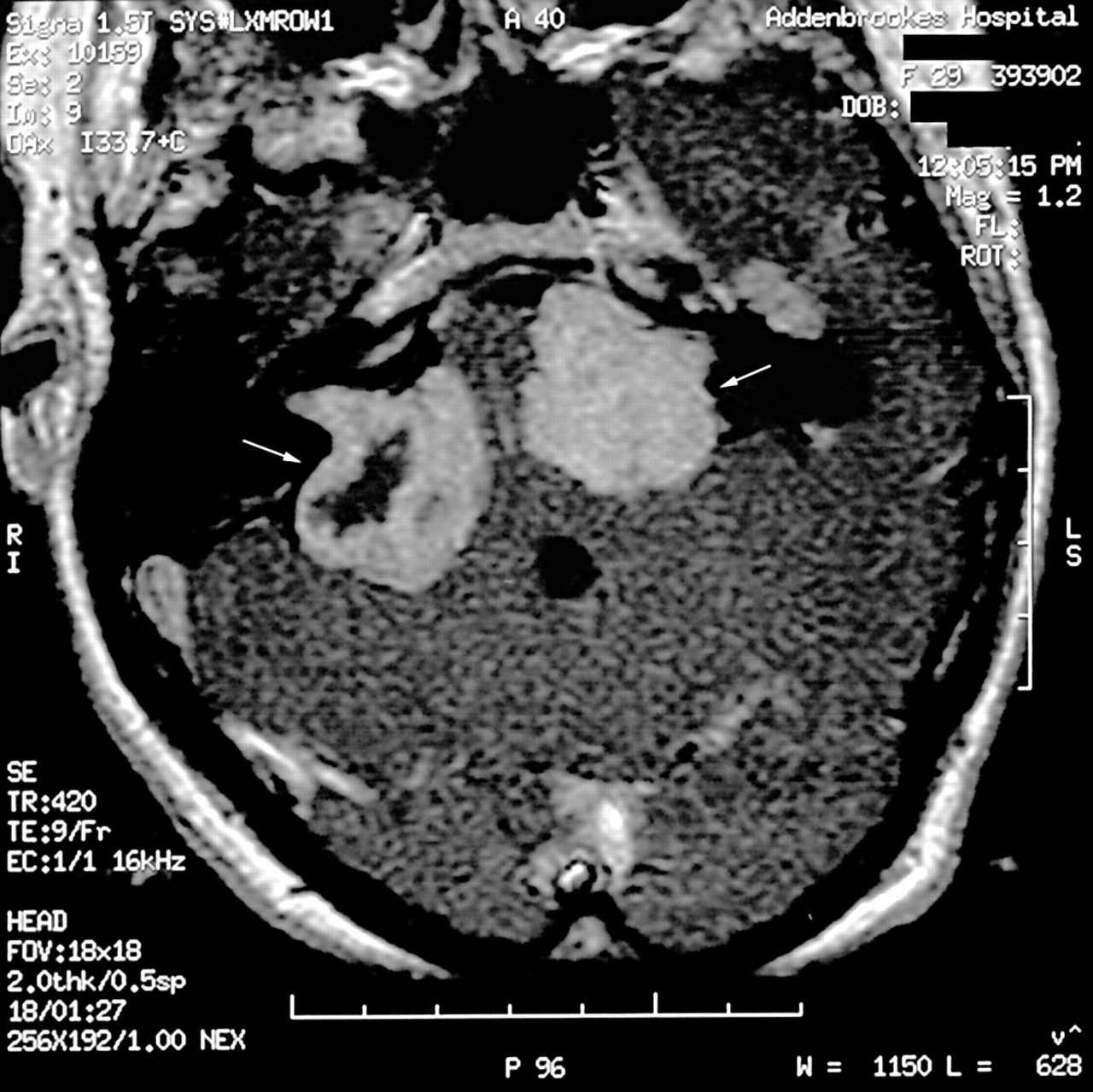
The diagnosis of NF2 was suggested in the patient once the histology of the abdominal schwannoma was identified. The diagnosis was confirmed when bilateral vestibular schwannomas were identified on magnetic resonance imaging (MRI) of the brain (fig 1). Initially she did not report pain or loss of hearing; however, as the tumours progressed she developed progressive hearing loss, ataxia, visual disturbance, and dysphagia over the subsequent three to four years.
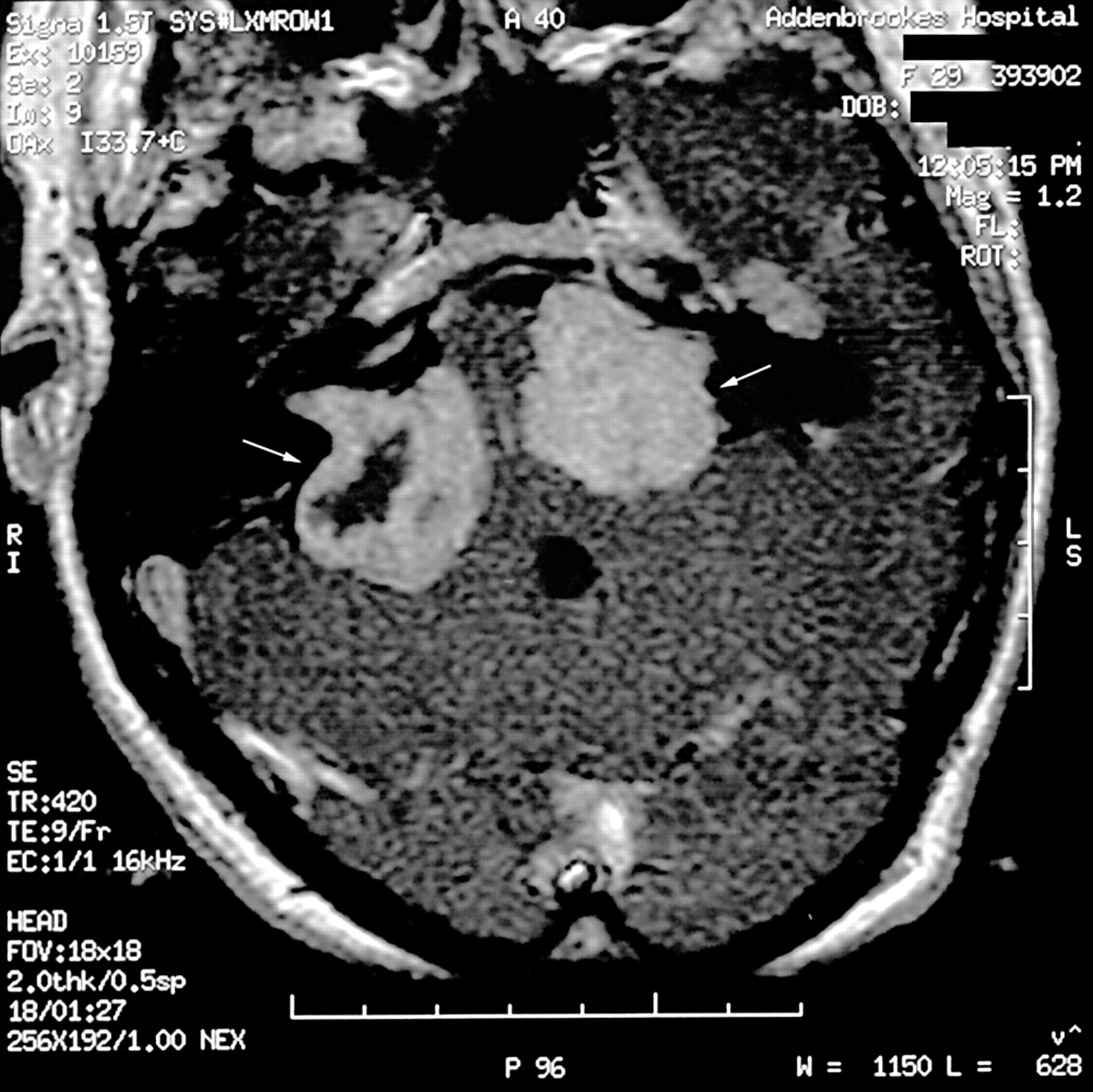
Magnetic resonance imaging of the head showing the presence of bilateral vestibular schwannomas, using a T1 weighted Gadolinium enhanced axial image. The vestibular schwannomas are indicated in the figure by white arrows.
Key points
-
This is the first reported case of neurofibromatosis 2 in a patient with a balanced X;22 translocation.
-
The chromosome 22 translocation breakpoint was 6 Mb centromeric to the NF2 gene and was defined by a single breakpoint spanning cosmid. No mutations or deletions were found in the germline NF2 gene of the patient.
-
The X inactivation pattern in lymphocytes was 100% skewed to inactivate the normal X chromosome as predicted for X;autosome translocations whereas in tumour tissue, there was aberrant X inactivation of the opposite derivative X chromosome.
-
The likely mechanism of disease in this case is that a proportion of Schwann cells have one NF2 allele acting as a functional null by virtue of NF2 being translocated to the X chromosome and aberrant X inactivation of the X;autosome.
Chromosome analysis of a cultured blood sample was done to identify a common aetiology for the presence of intellectual disability and a diagnosis of NF2. This revealed a constitutional karyotype abnormality, 46,X,t(X;22)(p11.2;q11.2). This had arisen de novo, as both parental karyotypes were normal. There was no evidence of NF2 in either parent, based on clinical examination and MRI investigation for bilateral vestibular schwannomas. The initial hypothesis was that the translocation involved the NF2 gene, with additional loss of chromosome material that would account for her intellectual disability. FISH analysis using PAC RP1-76B20 (AC004882), which contains most of the genomic sequence of NF2, showed that no such gene disruption or deletion was present (fig 2).

Fluorescence in situ hybridisation (FISH) using PAC RP1-76B20 (AC004882) containing genomic sequence from NF2 gene (green) and cosmid M0531 from chromosome 22 (red). The 22 breakpoint spanning cosmid is seen as red signal on chromosome 22, der(22) and der(X), and lies away from the intact translocated NF2 gene on 22 and der(X) (green). The distance between the breakpoint spanning cosmid and the NF2 gene is 6 Mb.
In addition, no mutation in the coding sequence of NF2 was identified by sequencing all 17 exons. Similarly, the presence of a smaller intragenic deletion was excluded by using Southern blot analysis of the whole genomic region in fragments.8
We identified the translocation breakpoint on the derivative chromosome 22. This was within BAC clone CTA-295D5 (B18082). Unique sequences from the ends of this BAC were identified and used to screen a chromosome 22 specific cosmid library, LLNL22NCO3, by PCR. Only one cosmid, M0531, contained both sequence homology to BAC CTA-295D5 and also spanned the chromosome 22 breakpoint by FISH (fig 2). This defined the breakpoint sequence to be within a 40 kb interval. The approximate location of the chromosome 22 breakpoint is at position 20 560 K on chromosome 22 as the sequence from the breakpoint spanning BAC, CTA-295D5, overlaps with clone KB-1572G7 (AP000346), which can be found on <http://www.ncbi.nlm.nih.gov/mapview/>. This is approximately 6 Mb from the genomic location of the NF2 gene which lies at position 26 720 K on chromosome 22 (Y18000). The translocation appeared balanced at the resolution of a single cosmid but the exact nucleotide sequence at the breakpoint was not determined. Known genes at the translocation breakpoint region are the immunoglobulin λ-like polypeptide 1 gene (IGLL1) and a hypothetical gene LOC51233. There are also several UniGene clusters in this region: Hs. 370433, 348935, 272302, 350074, and 350465. Further analysis of the breakpoint sequence contained in cosmid M0531 on chromosome 22 was not undertaken. This would be required to elucidate whether or not a novel or known gene has been disrupted at the chromosome 22 translocation breakpoint.
We then analysed the level of X inactivation from both the patient’s blood and tumour tissue removed from the abdomen to identify differences. Using a methylation dependent assay at the AR locus we could distinguish both X chromosome alleles.56 In blood the X inactivation pattern was skewed 100%, in keeping with the predicted arrangement for X;autosome translocations (fig 3, lanes 1–4).10 In tumour, however, the X inactivation pattern showed skewing in favour of the opposite allele, suggesting that the derivative X chromosome carrying the translocated copy of NF2 was being inactivated in a significant proportion of cells from the tumour (fig 3, lanes 5–8).

{kind=link}
{kind=link}
{kind=link}
X inactivation study of blood and tumour from the patient. Methylation status of the polymorphic human androgen receptor gene at Xq11-12 was assessed. HpaII selectively cuts unmethylated DNA, while McrBC only cuts methylated DNA. The polymerase chain reaction (PCR) products from each allele are marked A and B. On 9% denaturing PAGE these run as doublets. This shows an X inactivation pattern in blood (lanes 1–4) that is altered and skewed to the opposite X chromosome in tumour (lanes 5–8). B, blood; H, HpaII digested sample; M, McrBC digested sample; T, tumour; U, undigested sample.
DISCUSSION
Our patient presented with a large abdominal schwannoma and intellectual disability. This led to further investigations to identify a common aetiology. A clinical diagnosis of NF2 was made when bilateral vestibular schwannomas were found on MRI. With the demonstration of a de novo balanced reciprocal translocation between chromosome X and 22, we assumed that the disease in this patient was caused by the loss of NF2 at the translocation breakpoint. This assumption was incorrect and further investigation of the molecular basis of disease in this individual revealed an unusual but important mechanism of disease causation—namely, differing X inactivation patterns in different tissues. X inactivation in females is usually random. However, in patients with balanced X;autosome translocations the normal X chromosome is usually inactivated in order to preserve the expression of chromosome material from the autosome that has been translocated.9 In this patient, although the predicted pattern of skewed X inactivation was observed in her lymphocytes, we were fortunate in being able to extract DNA from the abdominal tumour. In tumour, a detectable proportion of cells showed inactivation of the opposite X chromosome. In these cells the NF2 allele that is translocated to the der(X) and lies only 6 Mb from the breakpoint is likely to be silenced as the X inactivation signal spreads into the autosomal region on the der(X). This means that, although the patient does not have a germline mutation to account for her NF2 disease, a proportion of her Schwann cells have one NF2 allele that is acting as a functional null by virtue of NF2 being translocated to the X chromosome and undergoing aberrant X inactivation. This would account for the relatively early age of onset of disease in this patient and the clinical severity similar to that seen in familial cases. It may also account for her intellectual disability if further genes from chromosome 22 had also been inactivated when translocated to the X chromosome in a subpopulation of brain tissue during early brain development.
This mechanism of disease is rare and has not been described before for NF2, although it has been described in cases of retinoblastoma.10–16 Mechanisms such as this should be considered when more classical mechanisms of disease have been excluded and where additional clues of accompanying intellectual disability are present.
Acknowledgments
This work was supported from the Addenbrooke’s Research Charity and the Wellcome Trust. STH is a Wellcome Trust clinical training fellow. We thank the patient and her family for their support and assistance in this work. We also acknowledge Mr D G Hardy, Mr D A Moffat, Dr N M Antoun, and Dr J R Anderson for their clinical involvement.