Article Text

Statistics from Altmetric.com
Williams-Beuren syndrome (WBS, OMIM 194050) is a microdeletion syndrome caused by hemizygosity for multiple genes in 7q11.23 including the elastin locus (ELN).1,2 The classical WBS phenotype comprises elastin arteriopathy (supravalvular aortic stenosis and/or peripheral pulmonary stenosis), connective tissue abnormalities (for example, abnormal joint mobility, hernia and diverticula, hoarse voice), and a particular facial appearance (supraorbital fullness, stellate pattern of the iris, short nose with long philtrum, full lips, and wide mouth). Other frequent features are growth and psychomotor retardation with muscular hypotonia, limited visuospatial cognition, and specific language as well as behavioural abnormalities (overfriendliness and anxiety disorders, hypersensitivity to sounds).3–5 Endocrine and metabolic disturbances (infantile hypercalcaemia) may occur.
Despite at least 21 genes having been identified in the common 1.5 Mb deletion interval in humans,6 their individual contribution to the multisystem phenotype of WBS is unclear. So far, only the gene coding for elastin (ELN) has been proven to be causally involved7: ELN is deleted in all WBS patients with a microdeletion 7q11.23 who have been reported to date. However, hemizygosity for ELN alone does not cause WBS, but isolated supravalvular aortic stenosis (SVAS).8 Hence, haploinsufficiency for ELN is necessary but not sufficient for WBS.
Molecular dissection of the WBS phenotype is hindered by the fact that over 95% of patients with the classical phenotype carry an apparently identical ~1.5 Mb deletion interval.9–11 This constant size is explained by non allelic-homologous recombination between duplicons flanking the deletion interval.12 The presence of these direct repeats is assumed to be a predisposing factor for the WBS microdeletion that occurs with a frequency of approximately 1 in 20 000 liveborn children.12
So far, several cases of either classical WBS or SVAS with or without cognitive deficits have been found to be associated with a smaller deletion. In seven of these cases an alternative proximal breakpoint was involved, and an eighth case featured an alternative distal breakpoint13–16 (reviewed in Osborne2). We present a further independent case with an atypical deletion but with the classical WBS phenotype. Together, these cases indicate that the critical WBS region is smaller than 1.5 Mb.
Key points
-
We present the case of a patient with the classical phenotype of Williams-Beuren syndrome who has only a partial deletion of the common 1.5 Mb deletion interval at 7q11.23.
-
Fluorescence in situ hybridisation (FISH) with the commercial WBSCR probe (from Appligene/Oncor) showed two specific signals in each metaphase, but repeat FISH with a second commercial FISH probe (LIS ELN from Vysis) showed a hemizygous microdeletion at 7q11.23. Additional FISH probes confirmed the partial deletion with the rare proximal breakpoint between STX1A and ELN and the distal common breakpoint around D7S489A.
-
We review published reports and discuss the implications of partial deletions for the diagnostic procedure and the correlation of genotype with phenotype in Williams-Beuren-syndrome.
CASE REPORT AND METHODS
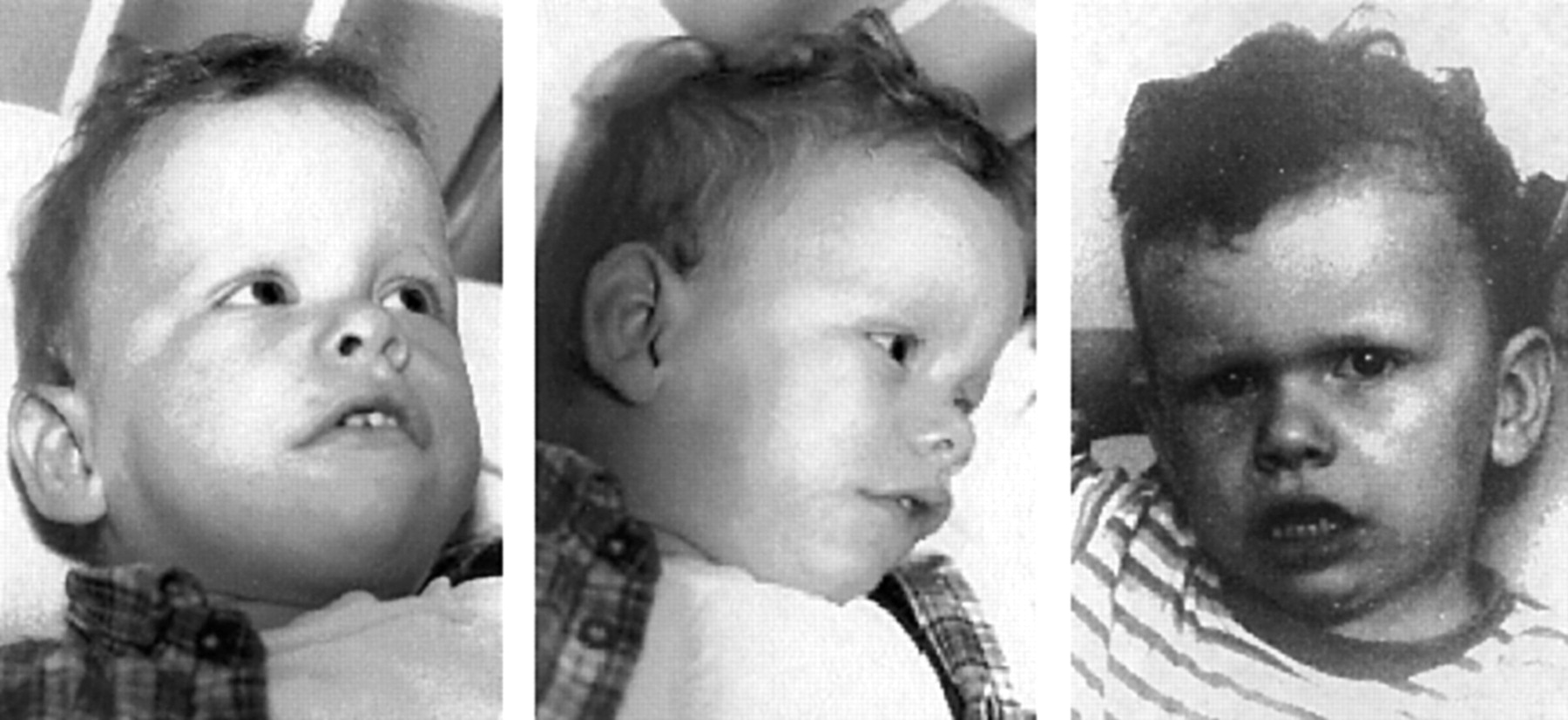
The patient and his healthy twin sister were born to non-consanguineous parents (maternal age 26, paternal age 41) after an uneventful pregnancy. Birth weight, height, and head circumference were within the normal range for twins. The constellation of supravalvular aortic stenosis, pulmonary valve stenosis, developmental retardation (not talking or walking at the age of 18 months), very friendly nature, and characteristic facial features in the patient led to the clinical diagnosis of Williams-Beuren syndrome (fig 1). Serum calcium levels from repeat measurements were within the normal range with one slightly raised value. When specifically asked about any behavioural traits, the patient’s parents mentioned his hypersensitivity to environmental noise in comparison to his healthy sister.

Patient with partial deletion of the Williams-Beuren syndrome critical region. Craniofacial features at 18 months (left) and 30 months (right) are compatible with Williams-Beuren syndrome: wavy hair, supraorbital fullness, upturned nose, full cheeks and lips, tented upper lip, and wide mouth. There is slight coarsening of the features with age, and the patient’s face at 30 months looks older than his age.
Chromosomes were analysed by conventional GTG banding of 15 metaphases from lymphocytes at approximately 500 bphs. For FISH analysis, commercial WBS probes from Qbiogene Alexis (catalogue No CP5155-DC) and from Vysis (catalogue No 32-190041) were used. Some of the non-commercial FISH probes were kindly provided by Dr Lucy Osborne, Department of Genetics and Genomic Biology, Hospital for Sick Children, Toronto, Canada.17–19 Labelling, hybridisation, and detection were performed according to the suppliers’ instructions and as described elsewhere.20
RESULTS
Initial chromosomal analysis yielded a normal 46,XY karyotype. FISH analysis with the commercially available WBSCR probe from Qbiogene-Alexis (catalogue No CP5155-DC) showed two specific signals at 7q11.23 in all analysed metaphases (example shown in fig 2A). However, there was a reproducible difference in signal intensity for the cognate probe (fig 2A) that was not found in the metaphases from either of the parents (data not shown). In view of the clinical diagnosis and of the fact that the WBSCR probe represents three different regions (around FZD9, LIMK1, and CYLN2) and covers almost the complete WBS chromosome region (fig 2A), a further 25 metaphases were analysed with the commercially available FISH probe from Vysis (catalogue No 32-190041). This probe consists of an approximately 180 kb probe covering the ELN and LIMK1 genes. There was one signal only in each of the 25 metaphases (fig 2B). The result was compatible with a de novo rearrangement that has led to a deletion including the ELN locus but clearly not of all the loci within the WBSCR that are recognised by the 7q11.23 WBSCR probe from Appligene-Oncor. We concluded that the WBS phenotype in this patient was most likely caused by an atypical 7q11.23 deletion.

Fluorescence in situ hybridisation with commercial microdeletion probe sets for 7q11.23 on metaphases from peripheral lymphocytes of our patient. Sequence contents as given by the manufacturers (Qbiogene-Alexis, Vysis). (A) No apparent deletion with the WBSCR probe, but different strength of signal for the cognate probe (white arrows). (B) Hemizygous microdeletion with ELN probe (missing signal marked by curved arrow).
Microsatellite analysis of DNA from peripheral blood lymphocytes of both parents and the patient showed biparental pattern for D7S2476 and DS489B, but was not informative for D7S613 and D7S1870 (data not shown, for location of markers see fig 3).

{kind=link}
{kind=link}
{kind=link}
Schematic diagram of the WBSCR in 7q11.23 with markers and genes indicated in the lower half. Modified from figure 1 in Osborne et al.18 The capital letters A–C above thick horizontal arrows indicate duplicated sequences and (pseudo-)genes that are arranged in three complex duplicons, one centromeric and two telomeric to the 1.5 Mb common deletion interval. The rectangles and circles above the diagram symbolise the FISH probes used in this study.17–19 Open symbols represent loci deleted on one chromosome 7, filled symbols stand for sequences present on both chromosomes 7. The distal breakpoint is defined by almost complete deletion of sequences that are recognised by PAC RP5-1186P10. Representative microscopic images are given at the top. The control probe in (A–D) contains chromosome 7 specific centromeric alphoid DNA and is labelled with rhodamine (red signals). The experimental probe is FITC labelled (green signals). In (E) the control probe at 7pter is labelled with FluorX (green signals) while the experimental probe is labelled with Cy3 (red signals). Note that the signal intensities for PAC clones H_DJ0953F13 and RP5-1186P10 differ in strength between the two homologous chromosomes 7, suggesting partial hybridisation on one chromosome 7 (marked by white arrows). The proximal and distal breakpoints in our patient are therefore assumed within the regions spanned by these two PACs. At the bottom, the extent of the partial deletions of the WBSCR that have been published is summarised. Cases 1–3: this report and Botta et al.13 Cases 4–8: refs 14–16. Case 9: refs 16 and 27.
To map the 7q11.23 breakpoints in our patient more accurately, PAC and cosmid probes for FISH analysis were used in dual colour analysis together with chromosome 7 specific centromeric alphoid DNA as control. The FISH results (fig 3) proved a partial deletion, caused by a distal recombination event at the common telomeric breakpoint,21 as suggested by the hybridisation signals for the PAC clones RP5-1186P10 and RP11-204E14. A proximal recombination event occurred around a rare breakpoint which is located at the centromeric end of PAC clone H_DJ0953F13. Both chromosomes 7 were positive for the cosmid probe cos16g10 (STX1A), but only one signal was seen for the cosmid probe cos82c2 (ELN 5′). The proximal breakpoint therefore lies in a region between STX1A and ELN.
DISCUSSION
Williams-Beuren syndrome belongs to a group of genomic disorders that result from regional genomic architecture predisposing to recombination events. In the case of WBS, non-allelic homologous recombination between segmental DNA duplications flanking the WBS region in 7q11.23 leads to a ~1.5 Mb deletion. The finding that the relatively well defined WBS phenotype is associated with an apparently identical deletion interval in >95% of cases9–11 initially raised hopes that a molecular dissection of the phenotype would be feasible. The number of genes identified within the deletion interval has increased steadily and has now reached at least 216 (UCSC Genome Browser on Human November 2002 Freeze at http://genome.cse.ucsc.edu/). However, the understanding of their role in the WBS phenotype has remained unchanged since the ELN gene has been identified as the cause of the vascular malformations in WBS,7,8,22 mainly because of the invariant deletion size in WBS.
Naturally, but rarely occurring 7q11.23 deletion variants in humans may help to identify the genes involved by phenotype-genotype correlation studies (fig 3). The first such atypical deletions were reported by Botta et al.13 Two patients out of 50 with the classical WBS phenotype had an uncommon centromeric breakpoint between the STX1A and ELN gene, hence questioning the role of the proximal WBSCR in the WBS phenotype. The patient reported here is the third case and thus supports the idea that the dysmorphic, cardiovascular, and at least some of the behavioural features of the WBS phenotype are caused by haploinsufficiency for genes in the part of the 1.5 Mb interval that is telomeric to the STX1A gene (fig 3). These genes do not only seem to be sufficient but also necessary for the WBS phenotype as illustrated by six partial deletions involving either just the ELN ± LIMK1 genes or the two-thirds of the 1.5 Mb interval centromeric to the RFC2 gene (cases 5–9). In none of the six cases was there a full WBS phenotype. Rather, these patients had either an isolated SVAS or an SVAS in combination with some degree of cognitive deficits.2 Our conclusion is that the true WBSCR is more likely an ~950 kb interval containing possibly 12 genes from WBSCR216 on the centromeric side to GTF2I on the telomeric side. This hypothesis is important as it limits the number of genes that potentially determine the WBS phenotype and excludes STX1A as a possible candidate gene. The lack of STX1A mutations in five patients, who have WBS but no detectable microdeletion at 7q11.23, supports this idea.23
Nevertheless, we cannot completely exclude that the partial telomeric deletions cause a position effect on the neighbouring chromosome region and may mimic haploinsufficiency for the complete set of genes in the 1.5 Mb WBSCR. The idea of a position effect is discussed in the recent publication of a spontaneously occurring familial translocation with disruption of the ELN gene in intron 5. Several members of this family who carried an identical translocation presented variably with WBS, SVAS, or no recognisable phenotype.24 The analysis of the mouse genomic sequence that is orthologous to the human 7q11.23 region will promote our understanding of the genomic organisation and genomic function of genes in this interval.17,25,26
Alternatively, the contribution of some of the genes in the centromeric part of the 1.5 Mb interval to the WBS phenotype could be subtle. For example, some parts of the cognitive and behavioural phenotype may not be clinically obvious in early childhood which is when most WBS patients are phenotyped. WBS and SVAS patients with an atypical deletion should be closely followed up into adulthood in order to detect any potential phenotypic difference from patients with the typical 1.5 Mb microdeletion.27
As a lesson from our particular case, we would like to remind cytogeneticists that partial microdeletions can result in false positive FISH signals depending on the sequence composition of the FISH probe used. This was first put forward as a hypothesis by Robinson et al28 and is now illustrated by our case and the cases described by Botta et al.13 Partial deletions of the WBSCR, and indeed other microdeletion intervals, might be more frequent than assumed. Had we only used the ELN FISH probe, the atypical nature of the deletion in our patient would have gone unnoticed.
Acknowledgments
The authors wish to thank the family of the patient for their cooperation, Michaela Kirsch for technical assistance, and Dr L R Osborne (Toronto) for providing several FISH probes.