Article Text

Statistics from Altmetric.com
Neurofibromatosis type 1 (NF1) is an autosomal dominant disorder with an estimated birth incidence of 1 in 2500 and marked variability of expression. The hallmark symptoms of the fully manifested disease encountered in nearly all patients are cutaneous neurofibromas, café au lait spots, axillary freckling, and Lisch nodules. Other common manifestations are bone dysplasias, scoliosis, vasculopathy, and learning disabilities. NF1 patients also suffer from an increased risk of specific tumour types like plexiform neurofibromas, neurofibrosarcomas, optic gliomas, other CNS tumours, phaeochromocytomas, juvenile xanthogranuloma, and juvenile myeloid leukaemia. Mutations of the NF1 gene at 17q11.2 encoding neurofibromin are the molecular basis of the disease. Neurofibromin contains a GTPase activating domain and is a negative regulator of Ras GTPases. Homozygous inactivation of neurofibromin is associated with a dysregulation of Ras mediated signalling pathways and tumorigenesis in NF1 patients.1 More than 70% of the germline mutations are protein truncating and are distributed throughout the coding region.2–4 No distinct genotype-phenotype correlation concerning type and position of the mutations has been established, apart from patients with microdeletions of the NF1 gene region, which are associated with a more severe clinical phenotype and facial dysmorphism. This was recognised very early and confirmed by several studies.5–9 Molecular characterisation of the deletion boundaries showed that non-allelic recombination between two highly homologous sequences separated by ~1.5 Mb eliminates 14 genes together with the NF1 gene during germ cell development.10–14 These 60–85 kb spanning low copy repeats are derived from segments of the WI-12393 gene and contain sequences with homology to chromosome 19. The structure of the NF1 gene region at 17q11.2 is further complicated by other duplicated sequences, such as pseudogene exons of the SMURF2 and the KIAA0160 genes.10,15 Up to now, homologous recombination between these duplicated sequences during mitotic cell divisions has not been reported. Here, we describe two brothers with severe NF1. Their mother, who does not fulfil the diagnostic criteria for NF1, only has a few inconspicuous café au lait spots and lacks any neurofibromas and other tumours. She shows somatic mosaicism for a large deletion of the NF1 gene region in peripheral blood leucocytes and skin fibroblasts. We determined the mechanism underlying the deletion and the size of the deleted interval at the cytogenetic and molecular level with respect to the duplicated sequences in the NF1 region.
MATERIAL AND METHODS
Patients
The 60 year old female investigated in this study (II.2, fig 1) has four café au lait spots on her thighs and forearms. Some minor freckling was found in the left axilla, but she did not have dermal neurofibromas or Lisch nodules. Her two sons, however, suffer from a severe form of NF1 (patients III.1 and III.2). In early childhood, they had multiple café au lait spots distributed all over the body and noticeably large hands and feet. In the younger brother, retarded psychomotor development was documented. Later in life, coarse facial features with hypertelorism were noticed, in addition to macrocephaly, axillary freckling, mild scoliosis, and multiple neurofibromas, which were too numerous to count. Multiple melanocytic naevi and low set nipples were additional features. The intelligence quotient of the sons was judged by their mother as normal, but was specifically tested only in her oldest son and was found to be in the normal range. Both attended regular school. Patient III.1 suffered from a malignant glandular schwannoma in the left gluteus which metastasised. At the age of 29 years, he died from intracranial bleeding before genetic investigations were started. His younger brother, patient III.2, who is still alive, had sphenoid wing dysplasia. At the age of 11 years, progressive dislocation of his hips and genu valgum were diagnosed, which had to be surgically corrected several times.

Genotype analysis of 11 polymorphic markers flanking the NF1 gene region and two markers within the NF1 gene (D17S1849, D17S1166) in the family of the index patient III.2. The haplotypes of patient III.2 were determined by analysing two hybrids, Nos 4 and 15, carrying only the deleted chromosome 17 and two hybrids, Nos 8 and 17, with the normal chromosome 17. Patient III.2 is hemizygous for markers D17S635, D17S1849, D17S1166, and D17S1800. Markers in parentheses located within the deleted interval of proband II.2 are present as single copies in the majority of peripheral blood leucocytes.
Key points
-
Large deletions of the NF1 gene region at 17q11.2 have been observed in 5–20% of all NF1 patients and are most frequently about 1.5 Mb in size. In the majority of these NF1 patients, a segment of a similar size carrying 14 functional genes is eliminated during meiosis by homologous recombination between low copy repeats (LCRs) of the WI-12393 gene and sequences with homology to chromosome 19.
-
Here we report on two brothers suffering from a severe form of NF1 and their mother, who does not fulfil the general diagnostic criteria for NF1. Marker and FISH analyses showed mosaicism for a large microdeletion of the NF1 gene region in the mother. The microdeletion was transmitted to her younger son and probably to her older son as well, who died from complications of the disease some years ago.
-
By contrast to previously characterised microdeletions, we located the breaks in another duplicated segment of the NF1 region, the KIAA0160 gene and its pseudogene. The disrupted KIA0160 gene is closely flanked by the WI-12393 gene, which is retained, and by WI-12393 related LCR sequences on the distal and proximal side, which have been shown to be involved in homologous recombination in other patients with NF1 microdeletions.
-
We conclude that somatic recombination between the KIAA0160 gene, which has also been called JJAZF1, and its pseudogene caused gonadosomatic mosaicism of the large deletion in the mother. Furthermore, marker analysis showed that mitotic intrachromosomal recombination is the mechanism underlying this deletion.
FISH analysis
Chromosome spreads were prepared from peripheral blood lymphocytes and skin fibroblasts of proband II.2 and blood lymphocytes of her son (the index patient III.2) according to standard methods. Skin fibroblasts of the mother were obtained by a punch biopsy of the left axillary region. BAC clones used as FISH probes were purchased from the BAC/PAC Resource Center (www.chori.org/bacpac) and have previously been characterised.10,11,15 PCR products used as FISH probes, DJ1686/1863, DJHK10/11, and DJ1576/1578, were amplified with primers listed in table 1 using the Expand Long Template PCR System (Roche Molecular Biochemicals). The respective products were cloned with the TOPO TA Cloning System (Invitrogen) and labelled for FISH analysis. Two colour FISH was performed using BAC-DNA or the cloned PCR products labelled either with biotin-16-dUTP or digoxygenin-11-dUTP (Roche-Diagnostics, Mannheim, Germany). Hybridisation signals were visualised by FITC-avidin and biotinylated anti-avidin (Vector, Burlingame, USA) or with anti-digoxygenin antibodies and in a second step with anti-mouse antibodies conjugated to Texas-Red (Dianova, Hamburg, Germany). Slides were counterstained with diamidinophenylindole (DAPI) and mounted with Vectashield antifade solution (Vector, Burlingame, USA).
Oligonucleotides to amplify regional PCR products used as FISH probes
Genotyping and generation of hybrid cell lines
Markers analysed were D17S1873, D17S1841, D17S975, D17S1863, and D17S635 proximal to the NF1 gene and markers D17S1800, D17S1880, D17S907, D17S1833, D17S1788, and D17S1867 distal to the NF1 gene. Markers D17S1849 and D17S1166 within the NF1 gene were also investigated. Genotyping was performed with 6FAM labelled primers and capillary electrophoresis on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems). For PCR experiments, we used DNA isolated from blood lymphocytes and hybrid cell lines carrying the deleted chromosome of patient III.2 (hybrid line Nos 4 and 15) and from hybrids with the normal chromosome 17 of III.2 (Nos 8 and 17) to identify the haplotypes. The hybrid cell lines were generated by PEG mediated fusion of a mouse cell line with peripheral blood lymphocytes of patient III.2 (GMP Genetics, Suffolk, UK). Hybrid cell lines were genotyped with the standard set of markers routinely used by GMP Genetics. Marker analysis in all other members of the family was performed using genomic DNA from peripheral blood.
Analysis of markers within the genomic interval between the KIAA0160 gene and its pseudogene
Dinucleotide repeat marker IVS27AC28.4 in intron 27b of the NF1 gene was amplified with Cy5 labelled PCR primers as described by Lazaro et al16 and analysed on an ALF-Express Sequencer (Amersham Pharmacia). The single nucleotide polymorphism SNP2 located in the KIAA1821 gene was amplified with primers described by Eisenbarth et al.17 Resulting PCR products were digested with the restriction enzyme MboI and analysed by agarose gel electrophoresis.
Fine mapping of the deletion breakpoints
The region of the deleted interval and the adjacent segments were analysed by PCR using polymorphic and non-polymorphic STS markers and DNA from a hybrid cell line carrying only the deleted chromosome 17 of patient III.2. PCR products flanking the deletion boundaries, DJ1911/1910, DJ1561/1562, and DJ1948/1936, were amplified with primers DJ1911 (5′ CTGCACATGGCATTGTTTGATT 3′) and DJ1910 (5′ AAACCCTCCTTGTGCCCTACTC 3′), DJ1561 (5′ GTCCAATAAG CAAGAAAAGAGCAA 3′) and DJ1562 (5′ TGGGGAAGAAAACATCAGTCAA 3′), DJ1948 (5′ GAGAAAATGAAAGGAGAGCAAGAA 3′) and DJ1936 (5′ TCTTAGTGCCTCTGGGAGCAA 3′). PCR products were directly sequenced by cycle sequencing on an ABI 377 sequencer (Applied Biosystems) and analysed using the GCG software (Genetics Computer Group) to determine their origin and to evaluate whether sequences are derived from one or two distinct loci.
RESULTS
Deletion detection by haplotype analysis and FISH
Analysis of polymorphic markers showed a large deletion of the NF1 gene region in the index patient III.2, as hemizygosity for markers D17S635, D17S1849, D17S1166, D17S1800 was observed in the DNA of peripheral blood lymphocytes (fig 1). To confirm these findings, FISH was performed with BAC 142O6 (AC079915), which covers the proximal region of the NF1 gene.15 A deletion of the corresponding region on one chromosome 17 was observed in all blood lymphocytes of patient III.2 (n=50). Upon this finding, the mother (patient II.2) of the index patient was also investigated by FISH. In her blood lymphocytes, the deletion was detected in 70% of the metaphases analysed (n=50), whereas in fibroblast cultures, the deletion of BAC 142O6 was noted only in 15% of all metaphases (n=20) (data not shown).
Delineation of the deletion boundaries by FISH
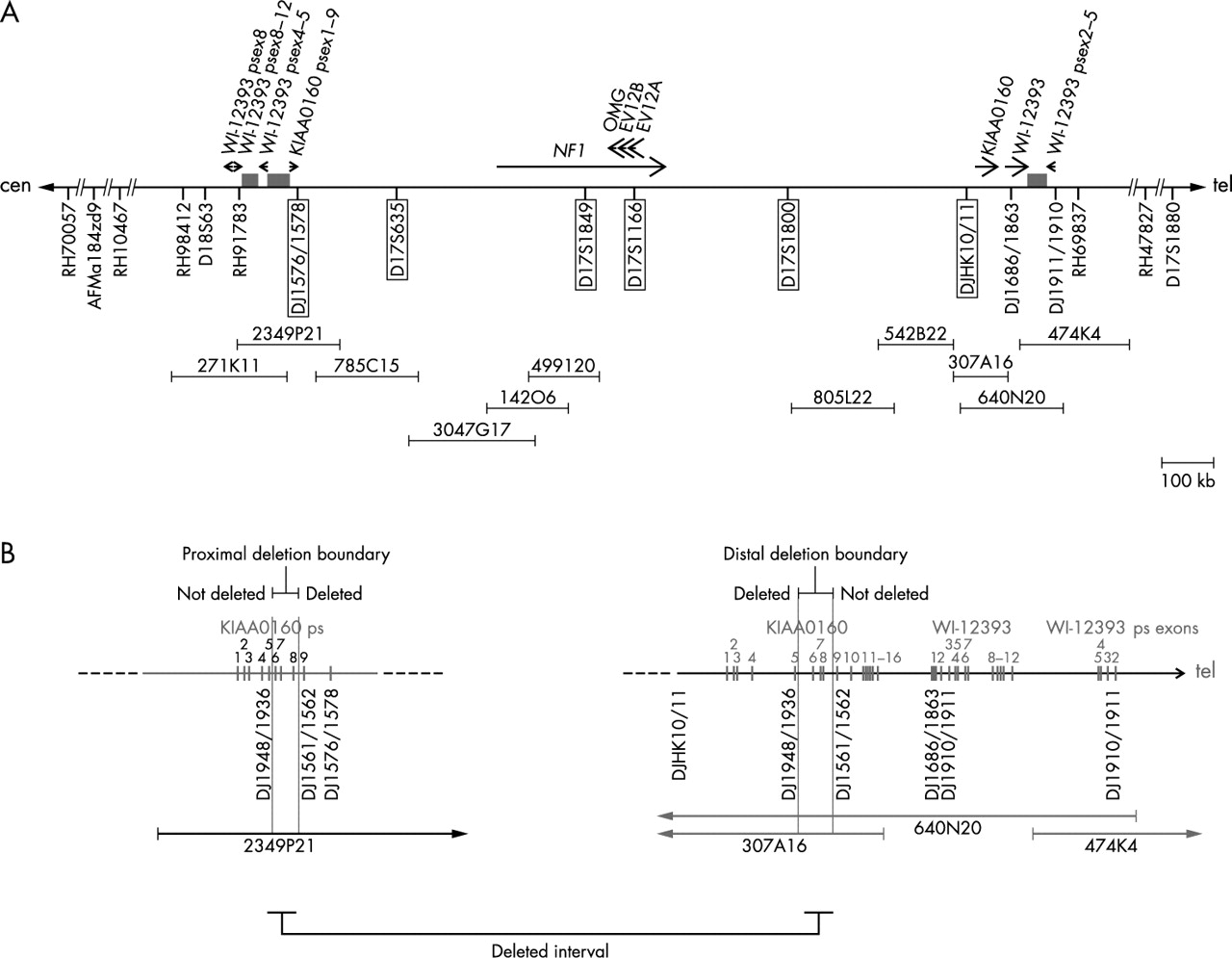
To determine the extent of the deletion, we performed FISH with further BACs that have previously been mapped to the NF1 gene region (figs 2 and 3).10–15 On metaphase chromosome spreads of the index patient III.2 and his mother, BACs 785C15, 3047G17, 499I20, 805L22, and 542B22 are deleted on one chromosome 17. Using BACs 2349P21 and 307A16 as FISH probe (fig 3A, B), a reduced signal was observed on one chromosome 17 homologue suggesting that these BACs span the deletion breakpoints on the affected chromosome. Convincing reduction of signal intensities was not observed using BACs 271K11 and 474K4 as FISH probes, which span the WI-12393 gene derived LCRs (fig 2A). To confine the deletion boundaries more precisely, FISH was performed with the cloned PCR products DJ1576/1578 amplified from BAC 2349P21 and DJHK10/11 amplified from BAC 307A16 (fig 2, fig 3C, D). Both probes are deleted on one chromosome 17 homologue. FISH probe DJ1686/1863, spanning exon 1 of the functional WI-12393 gene, hybridised to both chromosomes 17q11.2 and is therefore not deleted. These findings strongly suggest that the proximal deletion breakpoint maps to the region of the KIAA0160 pseudogene and the distal breakpoint to the functional KIAA0160 gene (fig 2B).

(A) Schematic presentation of the NF1 gene region at 17q11.2 and the localisation of STS markers as well as FISH probes. The deletion extent was determined by STS analysis of hybrid cell lines carrying only the deleted chromosome 17 from patient III.2 and by FISH on metaphase chromosomes prepared from the index patient (III.2) and his mother (II.2). The horizontal bar represents chromosome 17. The position of the NF1 gene, the functional KIAA0160 gene, the functional WI-12393 gene, and their pseudogene exons (psex) is indicated by arrows. STS markers and FISH probes (DJ1576/1578, DJHK10/11) are indicated by marks below the horizontal line. Deleted markers and probes are highlighted by a frame. BAC clones used for FISH are depicted by horizontal bars. The low copy repeats flanking the NF1 gene region consist of fragments of the WI-12393 gene and segments with high homology to chromosome 19 sequences, which are indicated by the grey rectangles. (B) Fine mapping of the deletion boundaries within the KIAA0160 sequences. The exons of the functional KIAA0160 and WI-12393 genes and their pseudogenes are shown by vertical rectangles with numbers based on the functional genes. The position of segments amplified with primers DJ1948/1936, DJ1561/1562, and DJ1910/1911 as well as the location of the FISH probes DJ1576/1578 and DJ1686/1863 are indicated beneath the horizontal line representing chromosome 17. As determined by FISH, PCR analysis, and sequencing of these products, the proximal deletion breakpoint maps between exons 5 and 9 of the KIAA0160 pseudogene. The distal deletion boundary is located between exons 5 and 9 of the functional KIAA0160 gene.

FISH analysis to investigate the deletion boundaries on metaphase chromosomes in the index patient III.2. BAC and PCR probes from the 17q11.2 region labelled with biotin and detected by FITC (green) were cohybridised with BAC 1D5 (red) as reference, which maps to 17p13. The BACs 2349P21 (A) and 307A16 (B) showed a reduced signal on one chromosome 17 (arrowhead) and thus cover the proximal and distal deletion boundaries, respectively. FISH analysis of probe DJ1576/1578 (C) and DJHK10/11 (D) showed the complete absence of the corresponding sequences on one chromosome 17 homologue marked by an arrow.
Identification of the deletion boundaries by PCR
PCR analysis of several STS using DNA from hybrid cell lines that carry only one chromosome 17 homologue of the index patient III.2 allowed us to confirm the presence of a large deletion (fig 2A). The markers that are framed in fig 2A were absent in the hybrid lines 4 and 15, but could be amplified from DNA of hybrids 8 and 17. To narrow down the boundaries more precisely, PCR products located within duplicated KIAA0160 and WI-12393 regions were amplified from the hybrid cell lines carrying the deleted chromosome 17 homologue and were sequenced. The location of these three primer pairs, DJ1948/1936, DJ1561/1562, and DJ1910/1911, is indicated in fig 2B. Sequence analysis of amplified products and comparison with the known sequences for 2349P21 (AC127024), 778K9 (AC023266), 307A16 (AC003041), and 640N20 (AC090616) enabled us to determine the presence or absence of the functional WI-12393 gene and the distally located WI-12393 pseudogene fragment, as well as the functional KIAA0160 gene and its pseudogene sequences. The PCR product DJ1910/1911 amplified from total DNA of the hybrid cell line 4 carrying only the deleted chromosome 17 contained sequences from both the functional WI-12393 gene and its distally located pseudogene. This result clearly indicates that the deletion does not include these regions and that the breakpoint lies centromeric to intron 2 of the WI-12393 gene. The DJ1910/1911 segment occurs only within the LCR sequence on the distal side of the NF1 gene and is not present in the proximal LCR.15 Examination of the PCR fragment DJ1948/1936, however, showed that this product was exclusively derived from the KIAA0160 pseudogene fragment located on BAC 2349P21. The sequence of the DJ1948/1936 product was unique and did not contain the functional KIAA0160 gene which on the other hand could be coamplified from the hybrids with the normal chromosome 17. By contrast, analyses of the PCR product DJ1561/1562 showed sequences that were solely amplified from the functional KIAA0160 gene, which is located on BAC 640N20 and 307A16, whereas the PCR product from the normal chromosome was mixed up with sequences from the pseudogene. Therefore, we conclude that the distal and proximal breaks are located within a duplicated segment between exons 5 and 9 of the KIAA0160 gene and its pseudogene, respectively.
Mechanism underlying the deletion
To determine the recombination mechanism resulting in the deletion, polymorphic markers were also analysed in the grandmother, I.2, of the index patient. Haplotypes were determined in patient III.2 using the hybrid cell lines 4,15 and 8,17 carrying only the deleted or normal chromosome of the index patient III.2. The phases of the haplotypes of the grandmother I.2 were inferred on the most parsimonious assumption that the haplotype of II.2 is not the result of a recombination event between grandmaternal haplotypes within the interval analysed (fig 1). According to our findings, intrachromosomal recombination between highly homologous KIAA0160 sequences was associated with the deletion.
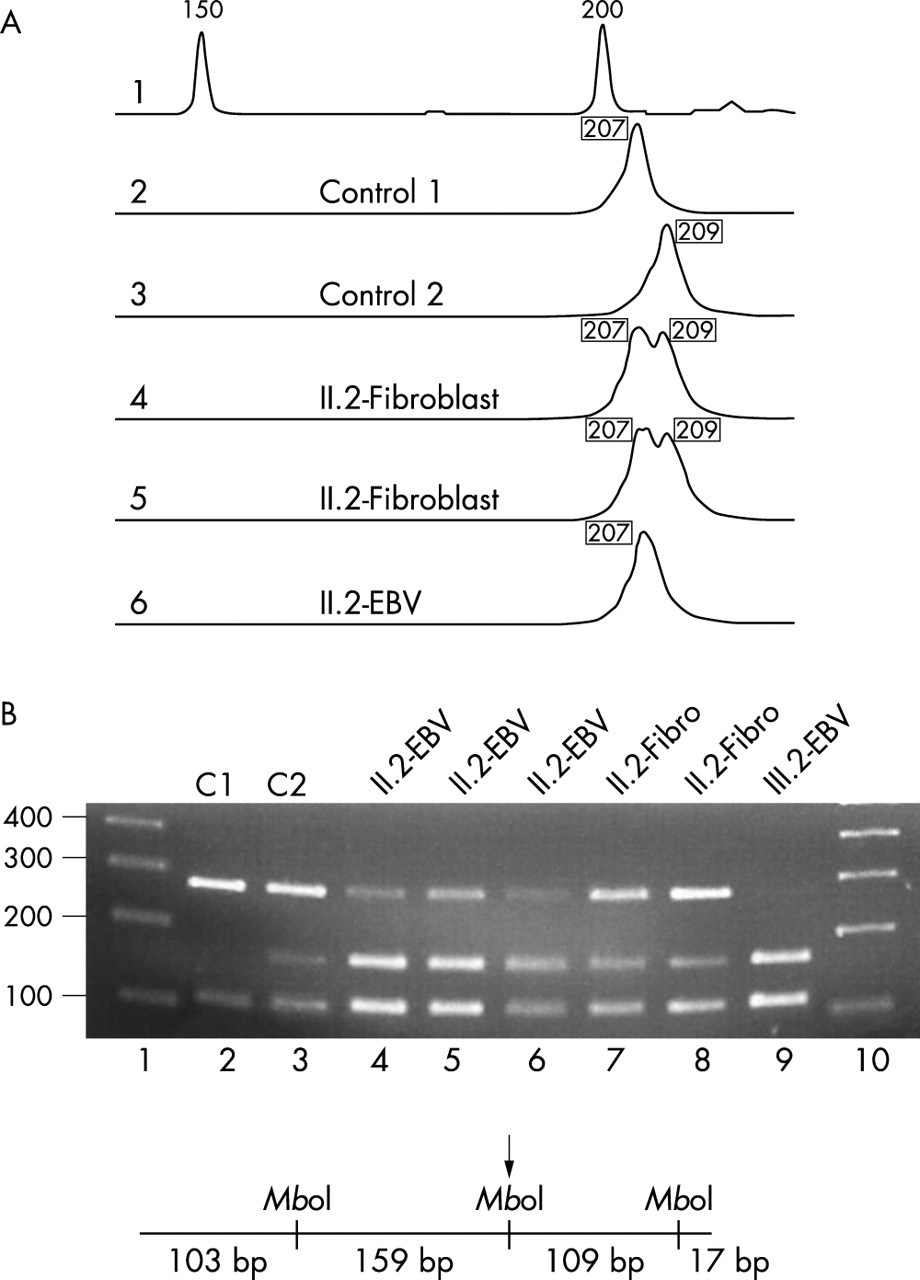
To show that the deletion occurred somatically in proband II.2, we investigated further markers in the genomic interval between the KIAA0160 pseudogene and the functional KIAA0160 gene and observed heterozygosity of proband II.2 in fibroblast cultures for the dinucleotide repeat marker IVS27AC28.4 in intron 27b of the NF1 gene and for the single nucleotide polymorphism SNP2 located in the KIAA1821 gene, flanking the NF1 gene in the 3′ direction (fig 4). The heterozygosity of these markers in normal cells of proband II.2 excludes the possibility that the germline deletion occurred during meiosis in the grandmother, and was partially corrected by mitotic recombination or segmental conversion of the deleted chromosome during early embryogenesis in somatic cells of proband II.2.

(A) Heterozygosity of the dinucleotide repeat marker IVS27AC28.4 located in intron 27b of the NF1 gene was observed in DNA isolated from primary fibroblast cultures. These cultures contained only a minor population of about 15% cells with del(17)(q11.2) (lanes 4 and 5, alleles of 207 and 209 bp). By contrast, the EBV cell line of patient II.2 mainly consists of cells (85–90%) carrying the deletion and thus only one allele of 207 bp was detected by this method (lane 6). In lane 1, an external size standard is given. (B) Heterozygosity of the single nucleotide polymorphism SNP2 in patient II.2. SNP2 is located in the KIAA1821 gene, which directly flanks the NF1 gene in the 3′ direction. The 388 bp spanning PCR product was digested with MboI before gel electrophoresis. As indicated in the diagram, the PCR fragment contains two constitutional MboI sites and one polymorphic MboI site (arrow). In lanes 1 and 10, standards are loaded with the band sizes given in base pairs on the left. Control DNA from C1 (lane 2) is homozygous for the allele lacking the MboI restriction site, the control C2 (lane 3) is heterozygous. In lanes 4 to 6 the MboI digested PCR fragments from patient II.2 were analysed and the results for different DNA preparations of the EBV cell line are shown. The percentage of cells carrying the deletion varied between 80–90% in different subcultures. Despite this high percentage of hemizygous cells present in the cultures, heterozygosity is observed at a disproportionate level. Most probably, single stranded DNA molecules without the MboI restriction site that are not extended during the final PCR cycles form uncleavable heteroduplexes with alleles containing the MboI site. These heteroduplexes presumably increase the amount of the uncleaved 268 bp band artificially. In conclusion, the fibroblasts of patient II.2 (lanes 7 and 8) are heterozygous, although they only contain 15% of cells with the deletion, whereas lymphocytes of the index patient III.2 are hemizygous for the marker SNP2.
DISCUSSION
The NF1 gene has one of the highest mutation rates in humans (~1 × 104/gamete/generation) and about 50% of all NF1 patients have no family history of the disease. This high mutation rate cannot be simply explained by the enormous size of the NF1 gene which spans 350 kb. It has been suggested that the high proportion of sporadic NF1 patients mirrors not only new mutations in the germline of a parent but also postzygotic NF1 gene mutations associated with gonadosomatic mosaicism in the transmitting unaffected parent (fig 5).18 Somatic mosaicism is clearly implicated in the pathogenesis of segmental neurofibromatosis,19 but only documented at the cytogenetic level for one patient.20 With regard to point mutations, somatic mosaicism of the NF1 gene is difficult to detect and to analyse in tissues, but may be one of the factors contributing to the relatively high number of sporadic cases and to the considerable variability of clinical signs and symptoms in NF1 patients.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The different origins of sporadic forms of NF1.
Interstitial microdeletions, however, can be approached by FISH more easily and thus somatic mosaicism has been detected in a few NF1 patients at the cytogenetic level.8,20–26 Large deletions of the NF1 region have been reported in six patients who all showed generalised NF1 symptoms not restricted to a particular body region. Four patients had deletions of the whole NF1 gene as shown by FISH analysis23–26 and two were ascertained by marker analysis.21,22 Moreover, Tinschert et al20 described a patient with segmental NF1 who is mosaic for an interstitial NF1 deletion. Breakpoint boundaries in all these seven cases have not been analysed so far and thus the underlying mechanism of mitotic rearrangements remains unclear.
Here, we describe a female with minor signs of the disease, who, however, is mosaic for a large deletion of the NF1 gene region. She transmitted this deletion to her son, who developed a severe microdeletion syndrome. The deletion encompasses a segment of ~1.3 Mb and is the result of non-allelic recombination between the KIAA0160 pseudogene on the proximal side and the functional KIAA0160 gene located distally to the NF1 gene (fig 2). This is the first published case of an NF1 microdeletion mediated by recombination within the KIAA0160 gene. In 29 of 60 NF1 patients with constitutional microdeletions characterised so far with respect to the deletion boundaries, the breakpoints were identified in highly homologous segments of the WI-12393 gene derived duplicons which flank the NF1 gene region.12–14 The KIAA0160 gene and its pseudogene are also duplicated segments, which are located in close proximity to the WI-12393 gene derived low copy repeats (fig 2A). The KIAA0160 gene, recently termed JJAZ1 (Joined to Juxtaposed with Another Zinc Finger gene) has been shown to be disrupted by somatically acquired translocations t(7;17)(p15;q21) in endometrial stromal sarcomas. This type of translocation not only fuses the promoter and 5′ end of the JAZF1 gene on chromosome 7 to the coding portion of the KIAA0160 (JJAZ1) gene on chromosome 17,27 but may also reduce the physiological activity of the KIAA0160 product as a result of hemizygosity at the KIAA0160 locus. While it seems premature to construct a link between hemizygosity of the KIAA0160 locus and tumour progression, it might be suspected that the triple event, loss of one JAZF1 allele, creation of a new fusion transcript between JAZF1 and KIAA0160, as well as the loss of one KIAA0160 copy triggers the development of endometrial sarcomas as a whole. Similarly, haploinsufficiency of KIAA0160 (JJAZ) may contribute to the outgrowth and progression of neurofibromas in microdeletion patients. Our speculation that KIAA0160 (JJAZF1) serves some tumour suppressive function, which may also be disturbed in endometrial stromal tumours, is consistent with the high risk and early development of tumours observed in patients carrying constitutional microdeletions.
The present study shows that the KIAA0160 gene in conjunction with its pseudogene represents a second recombinational pitfall in the NF1 gene region, besides the WI-12393 gene derived duplicons. Genotype analyses clearly indicate that the deletion occurred somatically in proband II.2 by an intrachromosomal (sister chromatid based) mechanism (figs 1 and 3). According to previous studies, low copy repeat mediated de novo deletions are believed to be triggered by meiotic recombination during germ cell development.14,28 This notion was based on PCR experiments that failed to identify the patient specific junction product in the blood of their healthy parents.14 López Correa et al28 observed a strong maternal bias for 17q11.2 microdeletions and found that the 1.5 Mb spanning deletions occurred predominantly during maternal germ cell development. In five of the six informative families, interchromosomal recombination (non-sister chromatid exchange) was the underlying mechanism that caused the rearrangements.
Our study implies that somatic recombination between duplicated sequences at 17q11.2 during early embryonic development can lead to gonadosomal and tissue mosaicism with only mild signs of NF1, which do not meet the diagnostic criteria. Subsequent germline transmission of such a microdeletion, however, can generate a very severe form of NF1 in the offspring with developmental retardation and high tumour load. The risk of somatic mosaicism in patients with very mild or ambiguous signs of NF1 who do not fulfil the usual diagnostic criteria is widely ignored or underestimated, but can have dramatic consequences, as in our family.
Acknowledgments
We thank Helene Spöri and Antje Kollak for expert technical assistance. The Institute of Medical Biology and Human Genetics at the University of Graz is a member of the IBMS and was supported by the infrastructure programme UGP4 by the Austria Ministry of Education, Science and Culture. This research was supported by grants of the Oesterreichische Nationalbank (No 9522/EP), by the FÖF (No S7403/PMK), and by the Deutsche Forschungsgemeinschaft (HA-1082 and KE-724 2-1/HK-S) and (KFO 113-1/DEJ).