Article Text

Statistics from Altmetric.com
A number of apparently non-syndromic X linked mental retardation syndromes are associated with subtle but characteristic phenotypic manifestations. Such manifestations can be dysmorphic features but they potentially also extend to abnormal brain morphology. In this latter field, progress in neuroimaging has aided the approach to brain malformations associated with mental retardation hence allowing a new classification of conditions previously described as non-syndromic. This classification is based on very similar brain malformations in affected subjects. Among the many brain malformations that can be associated with mental retardation in affected children, rhombencephalic anomalies are being recognised with increasing frequency. Accordingly, the classification of malformations of the posterior fossa has evolved considerably during the last decade.1–3
The cerebellum is known to be involved in movement coordination. However, besides its role in the control and integration of motor activity, the cerebellum also represents an essential node in the neural network subserving higher order behaviour.4,5 An abundant circuitry links the cerebellum with associative and paralimbic areas of the cerebral cortex and cerebellar lesions are known to underlie a cognitive syndrome combining impaired affective regulation, fine motor coordination, language fluency, verbal memory, and the ability to plan.4,5
These fascinating characteristics have led researchers to search for genetic determinants controlling cerebellar development. One way of addressing the genetics of cerebellar development in humans is to study families in which this brain region is abnormally developed. A number of families with X linked congenital cerebellar hypoplasia (CCH) have been reported,6–8 but no disease causing gene has been identified so far.
We have studied several families with X linked congenital cerebellar hypoplasia (CCH) and mental retardation and we have found different mutations in the oligophrenin-1 (OPHN1) gene. Carrier females are mildly affected and, accordingly, we found that they have a random X chromosome inactivation pattern in their lymphocytes. Our findings indicate that, in addition to its involvement in rare cases of non-specific X linked mental retardation,9 the OPHN1 gene plays a role during the development of the human cerebellum. This discovery also suggests that X linked inheritance may be relatively common in congenital cerebellar hypoplasia.
METHODS
Mutation screening
The OPHN1 gene was sequenced for each patient using genomic DNA as a template. A control sample (CEPH reference No 1347) was also sequenced. Primers were designed to amplify each exon of the gene (primer sequence available upon request). The PCR samples were directly sequenced, without subcloning, after purification with a Qiagen PCR purification kit. Sequence analysis and comparisons were performed using the Sequencher software (GeneCodes Corporation).
X chromosome inactivation studies.
Primers were designed in the (CAG)n flanking sequences of the androgen receptor (HUMARA) gene intron 1.10 Forward primer AR-P1 was 5′ labelled (IRD800) and the reverse primer AR-P2 was unlabelled. Primers sequences were: AR-P1, 5′ (IRD800)TCC AGA ATC TGT TCC AGA GCG TGC 3′; AR-P2, 5′ GCT GTG AAG GTT GCT GTT CCT CAT 3′. Four hundred ng of DNA were digested by HpaII and ethanol precipitated. PCR reactions were performed with 100 ng of DNA both on HpaII digested and undigested DNA for all the subjects. PCR conditions were as follows: 1 × PCR buffer, 0.2 mmol/l dNTPs, 1.25 Mg++, 0.5 U Taq (Gibco BRL) in 20 μl final volume. Annealing temperature was 60°C for 30 total cycles. The PCR product was diluted to 1/10th and directly loaded onto a LiCor automated DNA sequencer. The relative amount of each allele was scored using the OneD Scan software (Scanalytics).
RESULTS
Case reports (fig 1)

Pedigrees of families A and B.
Family A
Patient II.4, a boy, was born at term after an uneventful pregnancy. Birth weight was 3450 g (50th centile for age), length 51 cm (50th centile), and occipitofrontal circumference (OFC) 36.5 cm (90th centile). At the age of 9 months, he had febrile seizures. Generalised hypotonia and developmental delay were noted. Sitting was not achieved at 11 months. When he was 18 years old, he was referred to medical geneticists for diagnostic advice. Weight was 105 kg, height 1.82 m, and OFC 60 cm (>98th centile). The facial appearance was slightly dysmorphic with deeply set eyes and a prominent chin. Neurological examination was normal. He was significantly mentally retarded with no autonomy. Using the Wechsler Intelligence Scales, he was shown to have an IQ of 46. Reading was not acquired. MRI showed large ventricles without hydrocephalus but a blunted appearance of the angles of the lateral ventricles suggested a lack of white matter in the posterior fossa. There was a large cisterna magna without abnormality of the tentorium (fig 2A). Testing for fragile X syndrome was negative.
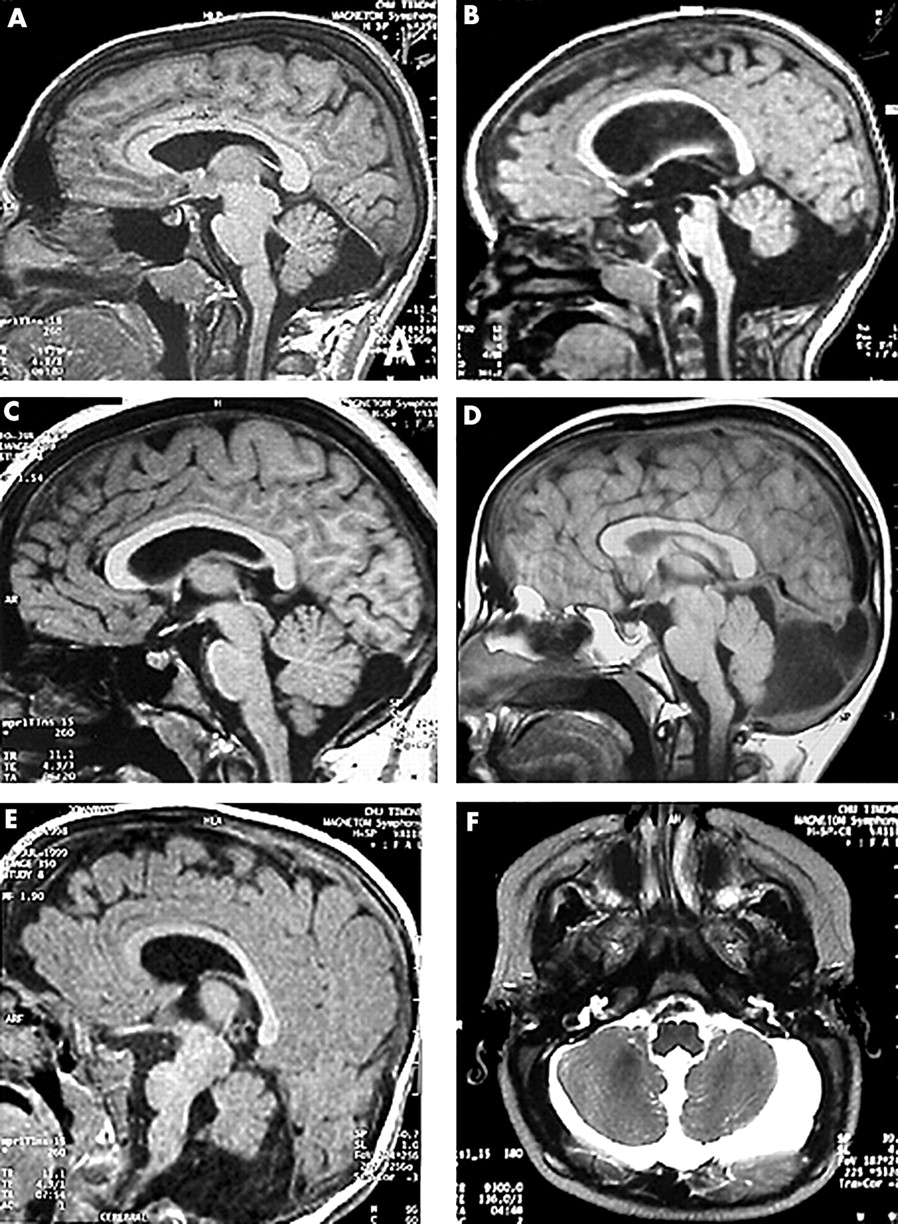
Magnetic resonance imaging results obtained for (A) family A, patient II.4; (B) family A, patient II.6; (C) family A, carrier mother II.2; (D) family B, patient II.4; (E, F) family A, patient III.2.
Key points
-
The classification of malformations of the posterior fossa has evolved considerably since high resolution magnetic resonance imaging (MRI) has become available. This technique has allowed researchers to address the genetics of cerebellar development in humans through the study of families in which this brain region is abnormally developed.
-
X linked congenital cerebellar hypoplasia (CCH) is one such condition. Although several pedigrees with X linked CCH have been reported, no disease causing gene has been identified so far.
-
We have screened six male patients (five familial cases and one sporadic case) with moderate mental retardation and cerebellar hypoplasia and have found two different mutations in the oligophrenin-1 (OPHN1) gene. Carrier females are mildly affected and we found that they have a random X chromosome inactivation pattern. The mutations are loss of function mutations disrupting the open reading frame of the OPHN1 transcript upstream of the known functional domains of the OPHN1 protein.
-
Our findings indicate that, in addition to its involvement in rare cases of non-specific X linked mental retardation, the OPHN1 gene plays a role during the development of the human cerebellum.
Patient II.5, the second brother, was born at term. OFC was 34 cm (50th centile), weight was 3800 g (75th centile). During the first months general hypotonia and macrocephaly were noted. At 11 months of age, OFC was 51 cm (>98th centile) and CT scan showed a marked dilatation of the lateral and third ventricles. In the posterior fossa there was a cystic expansion of the cisterna magna. This ventricular dilatation required ventriculoperitoneal shunt placement. Subsequent development was delayed and sitting was achieved at 15 months. On examination at 14 years of age, he showed macrocephaly (OFC 58 cm, 98th centile) and normal stature (161 cm, 50th centile). The facial appearance was very similar to his brother's. He had an IQ of 46. Supernumerary flexion creases were seen on the distal phalanges of his fingers. Mental retardation was severe, but without abnormalities on neurological examination. MRI showed large ventricles with a shunting device. In the posterior fossa the vermis was small with a large cystic cavity behind and around the cerebellum with a supratentorial expansion through a posterior hiatus of the tentorium. Fragile X syndrome was ruled out as it was in his older brother.
Patient II.6, is the half brother of patients II.4 and II.5. Second trimester amniocentesis was performed during pregnancy because of advanced maternal age. The fetal karyotype was normal 46,XY. Birth weight and length were in the normal range. OFC was 35.5 cm (60th centile). Generalised hypotonia and psychomotor delay were noted in the first months. At 3 years 3 months he was 100 cm tall (90th centile) and OFC was 53 cm (95th centile). Walking without support was acquired at 4 years of age. He had no language. Behaviour was abnormal with stereotypies and a tendency to self-mutilation. CT scan and MRI showed large, rounded lateral and third ventricles and a small vermis and cerebellar hemispheres. The posterior fossa formed a large cyst extending supratentorially though a posterior tentorial hiatus (fig 2B).
Patient III.2, was born at term. OFC was 36 cm at birth (90th centile). Psychomotor delay and peripheral hypertonia were noted in the first months. At 9 months of age, height was 75 cm (75th centile) and OFC 45 cm. Sitting was not acquired. MRI showed large ventricles, large pericerebral spaces, and poorly developed frontal lobes but without cortical dysplasia. In the posterior fossa the vermis was small with a large cisterna magna extending above the vermis and the cerebellar hemispheres.
Patient I.2, is the mother of three of the affected boys in family A. She was mildly mentally retarded. She had macrocephaly (OFC 60 cm) and her facial appearance was very similar to that of her sons. MRI showed a diffuse cerebral atrophy with a thick calvarium. No abnormality was found in the posterior fossa.
Patient II.2, is slightly mentally retarded and could not attend normal school. MRI showed a slight supratentorial expansion of the cisterna magna through a posterior hiatus of the tentorium (fig 2C). Two of her children, a girl and boy aged 1 and 3 years, had normal psychomotor development and MRI.
Family B
Patient II.4 was born at term to healthy, unrelated parents. Four sibs were healthy. Pregnancy and delivery were uneventful. Psychomotor delay was noted during the second year of life. Walking was achieved at 18 months of age and language was severely delayed. At 11 years of age, a brain CT scan was performed, showing a retrocerebellar cyst with tentorial dysplasia, a small vermis (fig 2D), and dilated ventricles requiring ventriculoperitoneal shunting. Clinical examination at 13 years showed dysmorphic features with deeply set eyes, prominent roof of the nose, and prominent chin. OFC was 57.5 cm (>97th centile), weight was 63 kg (>97th centile), and height was 158.5 cm (50th centile). Reading was poor and writing was not acquired. According to the parents, he had behavioural problems with instability and intolerance to frustration.
Linkage analysis and candidate gene selection
Since X linked inheritance was very likely in family A, we performed linkage analysis to map the disease locus. No significant lod score value was obtained but haplotype reconstruction and analysis of allele sharing between affected subjects showed that the region containing the disease locus was located between markers DXS1039 (Xp11.23) and DXS1047 (Xq25), excluding the major part of the short arm and the distal portion of the long arm (data not shown).
A recent report showed that some patients with complete androgen insensitivity and mental retardation syndrome (CAIS-MR) and large deletions in the Xq12 region also had cerebellar hypoplasia.11 Since the OPHN1 gene is located inside the deletion interval, since this interval is located inside the linkage region in family A, and since mutations in this gene are responsible for non-specific X linked mental retardation (MRX),9 it became a potential candidate gene for X linked congenital cerebellar hypoplasia.
Identification of mutations in the OPHN1 gene
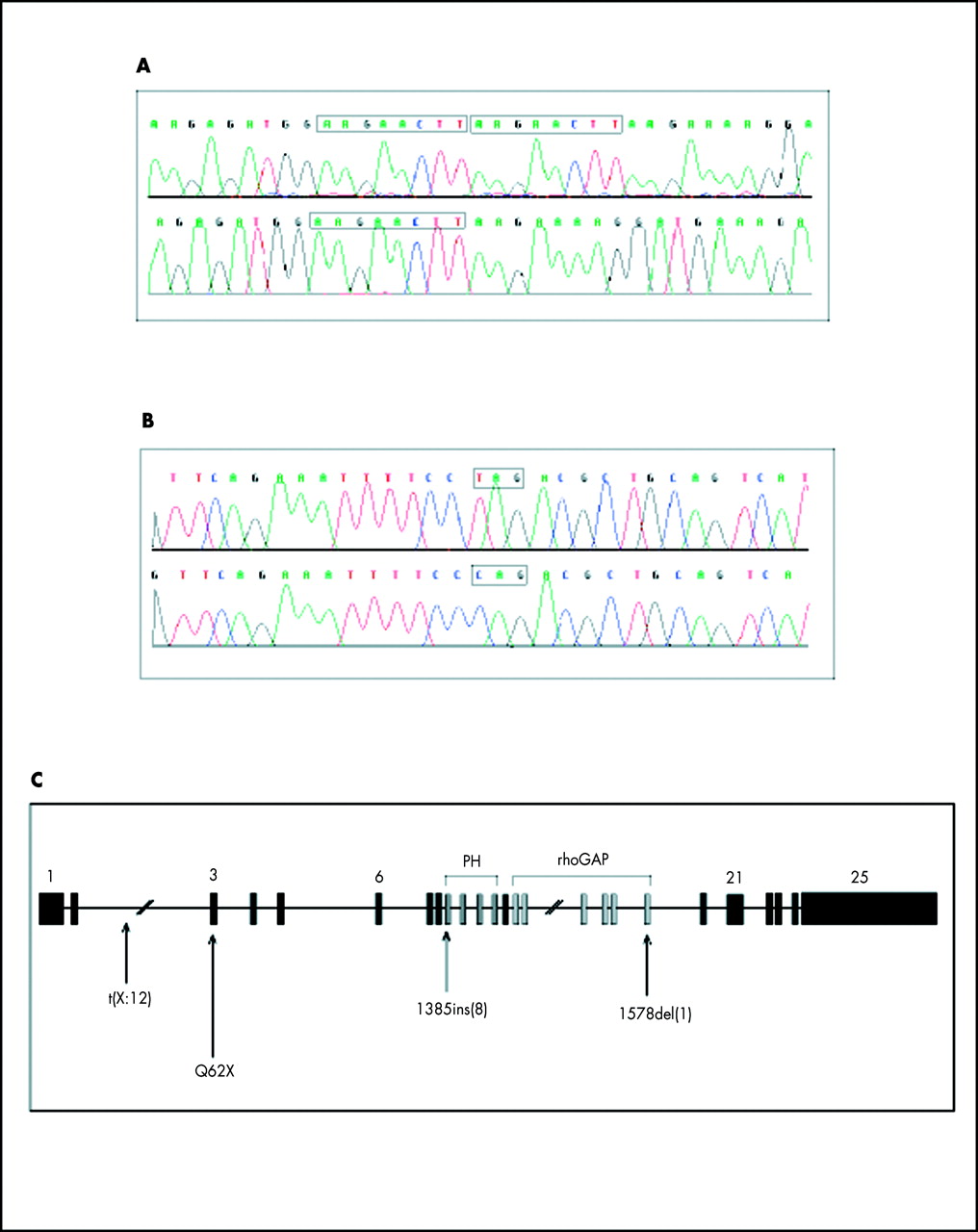
We sequenced the 25 exons of the OPHN1 gene in family A and found a duplication of 8 bp in exon 9 of the OPHN1 gene in all affected subjects and in their carrier mothers. This insertion causes a frameshift at nucleotide 1385 which, in turn, leads to a premature stop codon (fig 3A).

{kind=link}
{kind=link}
{kind=link}
(A) Electrofluorogram showing the 8 bp insertion (AAGAATTC) found in family A: top, patient II.2; bottom, control sample. (B) Electrofluorogram showing the C to T transition found in family B: top, patient II.4; bottom, control sample. (C) Schematic representation of the genomic structure of the OPHN1 gene. The 25 exons are shown together with the position of the coding region for the PH and the rhoGAP domains. Arrows indicate the t(X ;2) and 1578del(1) mutations found in MRX patients and the Q62X and 1385ins(8) mutations found in the present study.
In a further step, we selected five additional patients with a similar clinical presentation. Four patients originated from potentially X linked families where two males were affected with moderate mental retardation and cerebellar hypoplasia. The fifth patient is a sporadic case who was selected on the basis of striking similarities with the affected boys of family A: the same facial appearance with deep set eyes, prominent chin, large stature, macrocephaly, and identical MRI findings.
Screening of the OPHN1 gene showed that one patient (patient II.4 in family B) was also a carrier of a mutation in this gene. This patient has the same clinical and MRI features as affected patients in family A (fig 2D). He harboured a C to T transition in exon 3 which causes the replacement of the codon for glutamine 62 by a stop codon (Q62X, fig 3B). The OPHN1 protein contains two functional domains in its COOH terminal region, a PH and a rhoGAP domain. The PH (pleckstrin homology) domain binds membrane phosphoinositides.12 The rhoGAP domain is found in proteins transducing signals from membrane bound receptors and it is able to stimulate the GTPase activity of small G protein.13 In our patients, both mutations introduce a stop codon upstream of the region coding for these two domains (fig 3C).
We next wanted to determine if the OPHN1 transcript was present in normal quantity in the patient's cells although carrying a mutation. We performed RT-PCR experiments designed to amplify a 550 bp OPHN1 PCR product using the patients' mRNA as a template. We found that the OPHN1 transcript is present in the lymphocytes of all patients of family A in a quantity that is comparable to a control sample (data not shown). No mRNA sample was available to perform the same experiment in patient II.4 of family B and thus the presence of the mutated transcript was not tested in this case. These results indicate that, at least in family A, the mutated transcript is stable and that a truncated OPHN1 protein could possibly be produced upon translation of the mutated transcript.
X chromosome inactivation in carrier females
Since carrier mothers are affected, although to a lesser extent than their affected offspring, we studied their X chromosome inactivation (XCI) patterns. Circulating lymphocytes were used to prepare DNA and XCI was assessed at the androgen receptor (AR) gene locus (see Methods). We found that both carrier mothers in family A have a random XCI pattern in their lymphocytes (data not shown). No DNA was available for the mother of the patient in family B.
DISCUSSION
The phenotypic difference between the previous report of patients with a mutation in the OPHN1 gene9,14 and our patients is surprising. In family A, the most prominent manifestation is moderate to severe mental retardation. In all affected males, hypotonia and developmental delay were noted during the first months. Mean age at walking was 30 months. Language was severely delayed and the 18 year old boy had not achieved reading skills. In addition to mental retardation and macrocephaly, the only manifestations were tall stature and some degree of facial dysmorphism. Neurological examination was unremarkable apart from hypotonia. No symptom of patent cerebellar dysfunction, such as ataxia or dysarthria, was observed in any of the boys, in spite of their abnormal cerebellar morphology. On brain imaging, all subjects showed some degree of vermis hypoplasia and cystic dilatation of the cisterna magna, with posterior dysplasia of the tentorium. The size of the cisterna magna was measured with reference to a previous work on normal variations of the cisterna magna.15 Supratentorially, mild to large dilatation of the ventricles was noted without apparent cortical dysplasia. The two most severely affected boys (patient II.3 in family A and patient II.4 in family B) developed hydrocephalus, which required ventriculoperitoneal shunting.
Several pedigrees with congenital cerebellar hypoplasia displaying an X linked mode of inheritance have been reported. We compared the clinical manifestations of the family we describe with all previously reported X linked families. Details are provided in table 1. The first cases were published before the availability of MRI imaging. Riccardi and Marcus16 described two brothers who died shortly after birth with congenital hydrocephalus. Necropsy was performed in one and showed cerebellar agenesis. The family history, with the presence of hydrocephalus in a maternal great uncle was suggestive of an X linked mode of inheritance. In the other families, the neurological symptoms are variable (table 1). Concerning the MRI findings, it is worth noting that in poorly documented cases, when a single and late neuroimage is available, it can be difficult to differentiate hypo/aplasia from progressive atrophy. Linkage analysis was performed in some families. The fact that at least two families are linked outside the X chromosome region containing the OPHN1 gene means that cerebellar hypoplasia is likely to be a genetically heterogeneous condition. In addition, several of the previously reported pedigrees of X linked cerebellar hypoplasia were characterised by additional findings, which also may suggest genetic heterogeneity. The combination of hydrocephalus and cerebellar hypoplasia might suggest that the OPHN1 protein acts in the leptomeninges since dysplasia of the leptomeninges can lead to cerebellar hypoplasia and hydrocephalus.17
Summary of the previously reported families with X linked inheritance and similar clinical findings
The fact that we found two new OPHN1 mutations by screening a limited number of X linked CCH patients is rather unexpected. Indeed, only one OPHN1 point mutation has been published previously (besides the initial translocation patient) despite a considerable effort in screening males with non-specific X linked mental retardation.18 It is possible that the MRX patients found to harbour mutations in the OPHN1 gene are located at the “mild” end of a phenotypic spectrum associated with OPHN1 dysfunction and that they are relatively rare. According to this hypothesis, most mutations in this gene would cause congenital cerebellar hypoplasia and would currently be missed. A very large screen of all patients with CCH should thus be undertaken to answer this question. Given that carrier females are clinically affected, we suggest that both sexes should be included in this analysis. Families of children with congenital cerebellar hypoplasia or Dandy-Walker malformation are not generally counselled for a high risk of X linked inheritance. Our findings suggest that X linked inheritance may be relatively common in this condition.
Little is known about the function of the OPHN1 protein. In situ hybridisation experiments performed using the mouse OPHN1 transcript have been reported but no topographical details were given.9 The transcript is highly expressed in the mouse nervous system at different stages of development.9 Mutations in Rho GAP proteins may promote the constitutional activation of specific GTPases and subsequently affect cell migration and axon guidance, as was shown using the nematode C elegans.19 Moreover, some evidence has recently accumulate to implicate Rho GTPases in the regulation of neuronal morphogenesis.20 We believe that the recognition of the full spectrum of phenotypes associated with oligophrenin-1 dysfunction and appropriate functional studies will shed new light onto the processes leading to the development of the human cerebellum.