Article Text

Statistics from Altmetric.com
- DOA, dominant optic atrophy
- BCVA, best corrected visual acuity
- SNPs, single nucleotide polymorphisms
- LCNRs, low copy number repeats
Dominant optic atrophy (DOA) is the most common form of autosomally inherited optic neuropathy.1 The disease typically presents in childhood with slow bilateral loss of visual acuity, visual field defects, abnormal colour discrimination, and pallor of the optic discs. The majority of DOA families published to date have shown linkage to a major locus on chromosome 3q28 (OPA1). The OPA1 gene was recently identified and found to encode a ubiquitously expressed, dynamin related GTPase.2,3 In order to determine the mutation spectrum of OPA1 in DOA, we and others have screened the coding exons and their flanking splice sites in large patient cohorts.4–7 Over 60 different mutations have been reported, most of which are specific to individual families. It has been speculated that haploinsufficiency is the cause of disease,3–6 but to date there has been no evidence to prove that this mechanism, rather than aberrant function of mutated proteins, is responsible for the disease.
METHODS AND RESULTS
Using a combined approach of single stranded conformational polymorphism and heteroduplex analysis, we detected mutations in 57% (20/35) of our affected DOA patients.5 Additional DNA samples from an Australian family, in which no OPA1 mutation was identified in the above mutation screen, have recently been obtained enabling further investigation of the cause of disease in this family. All available family members underwent clinical evaluation to determine their disease status. This included best corrected visual acuity (BCVA), assessment of colour vision with Isihara plates and/or City University testing, automated perimetry (Humphrey Field Analyzer, San Leandro, CA), and dilated fundus examination with stereo disc photography. The ocular phenotype observed was typical of DOA with visual acuity reduction ranging from mild to moderate, mild to moderate colour vision disturbance, and visual field analysis ranging from normal to mild paracentral relative scotomas to more severe peripheral loss in some older patients. The optic disc appearance ranged from subtle temporal pallor to more generalised optic atrophy with or without disc excavation in some older patients.
We performed genotyping on genomic DNA from all available family members, using 12 microsatellite markers surrounding the OPA1 gene (fig 1, method described previously8). On first examination of the data, three of the markers (D3S1523, D3S3590, and D3S2305) appeared to be homozygous in every affected subject. However, the “homozygous” alleles differed among the affected subjects suggesting that affected persons were actually monoallelic at these loci. Thus, we had reason to suspect that this family has a chromosomal deletion at 3q28. In order to rule out non-paternity or any other possibility that could account for these results, each person was genotyped for microsatellite markers from chromosome 6 (ATA41H06, D6S284, D6S460, D6S1707, D6S391, and D6S445, data not shown). This allowed confirmation of Mendelian inheritance and paternity throughout the pedigree.
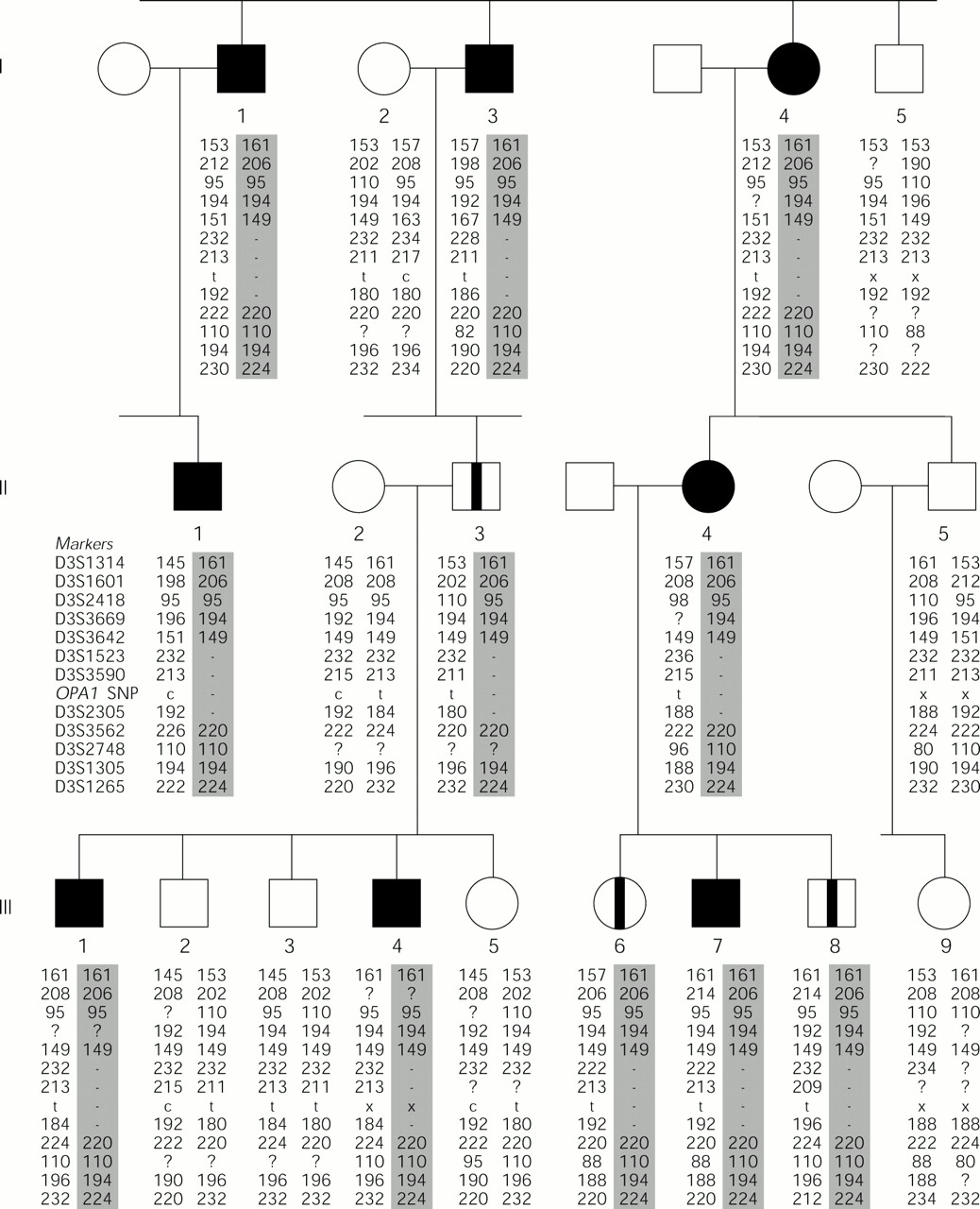
The partial pedigree of a DOA family with a deletion at 3q28 that segregates with the disease haplotype. Subjects were genotyped for 12 microsatellite markers surrounding the OPA1 gene (listed in order, proximal to distal) and three OPA1 intragenic single nucleotide polymorphisms (SNPs) (the results for SNP 557-19c/t are given). Haplotypes were assembled and the allele sizes in base pairs are given. Filled symbol = affected; open symbol = unaffected; open symbol with stripe = asymptomatic carrier; - = marker locus is monoallelic; x = marker not typed; ? = marker failed to amplify or unable to score peak.
Of the three deleted markers, two are located proximal to OPA1 and one is distal, implying that the deletion encompasses the full OPA1 gene. Further evidence was obtained by typing three OPA1 intragenic SNPs in family members. 557-19c/t, 870+4c/t, and 870+32t/c were sequenced following PCR amplification from genomic DNA. (Nucleotide numbering for SNPs follows the cDNA sequence (GenBank accession No AB011139), with position 1 assigned to the first nucleotide of the ATG initiation codon in exon 1.) The results obtained were in agreement with the microsatellite genotyping results as all affected subjects appeared to be “homozygous” for each SNP (data for SNP 557-19c/t are given in fig 1). It is noteworthy that the genotyping data for this family place two of the microsatellite markers (D3S1523 and D3S3642) in the reverse orientation to that in which they occur on physical maps of the region.8–10 D3S1523 is monoallelic in all affected family members while D3S3642 is not, implying that D3S3642 is proximal to the deletion and to D3S1523. Analysis of the sequence data currently available for this region could not resolve the apparent contradiction in marker order, owing to gaps in the human genome draft sequence for this region.
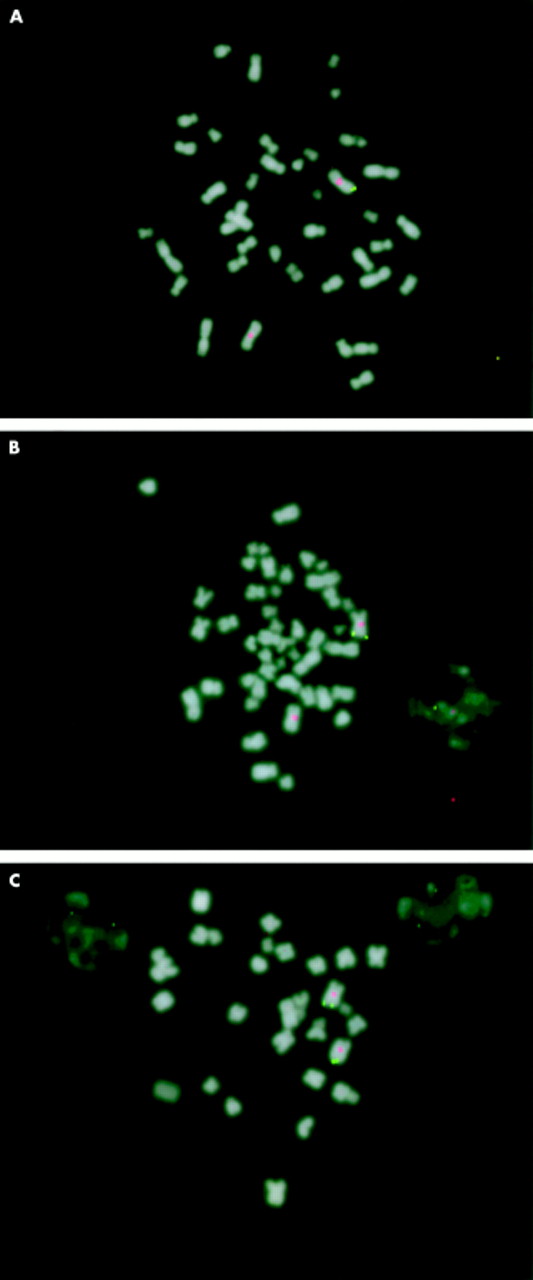
Fluorescence in situ hybridisation (FISH) analysis was performed in order to show that one copy of OPA1 is deleted in affected family members. OPA1 cDNA was cloned and used as a probe on metaphase chromosomes from an affected subject, an asymptomatic obligate carrier, and an unaffected spouse (fig 2, methods previously described11). The FISH results confirm the deletion of one copy of the OPA1 gene in the affected and the carrier subjects. Therefore, we conclude that haploinsufficiency of the OPA1 protein is the cause of disease in this family and, in all likelihood, in other DOA families with truncated or mutated OPA1 alleles.
{kind=link}
{kind=link}
FISH analysis showing the deletion of the OPA1 gene in a DOA family. Cloned OPA1 cDNA was used as a probe (green) along with a centromeric chromosome 3 probe (red) to indicate the absence of one copy of OPA1 in metaphase chromosomes from (A) an affected family member (I.3) and from (B) an asymptomatic obligate carrier (II.3). An unaffected subject (I.2) was analysed as a normal control (C).
Key points
-
We have identified a dominant optic atrophy (DOA) family with a 560-860 kb microdeletion on chromosome 3q28 that results in the complete loss of one copy of the OPA1 gene.
-
Our finding provides evidence that haploinsufficiency, rather than aberrant function of mutated proteins, is the cause of disease in DOA patients with mutations in the OPA1 gene.
-
Furthermore, this report raises the possibility that other cases of DOA in which no mutation was identified may have a deletion of the OPA1 locus.
DISCUSSION
Other examples of haploinsufficiency in human dominant disorders have been described in recent years. Not surprisingly, the causative genes are mostly involved in developmental pathways or participate in the assembly of intermolecular complexes.12 In either case, protein levels are crucial.
The size of the deletion in this family was estimated at 560-860 kb based on the physical distance between the microsatellite markers on our physical map.8 Small interstitial deletions are often the result of rearrangements in regions of the genome containing low copy number repeat units (LCNRs) that promote unequal crossing over.13 These types of repeat sequences have been found flanking the deletion breakpoints in a number of diseases such as Williams syndrome14 and DiGeorge/velocardiofacial syndrome.15 To our knowledge, there are no reports of LCNRs in this region. The deleted region in this family includes one other known gene in addition to OPA1, a Ha-RAS suppressor (HRASLS),16 and potentially up to nine other genes based on the estimated average human gene density of 9.6-12.9 genes/Mb.17 Indeed, we have previously mapped numerous expressed sequence tags derived from as yet unknown genes to this region.8
Since the deletion encompasses other genes in the 3q28 region, it was of particular interest to note the presence of pes cavus (high foot arch) and reduced deep tendon reflexes in an older affected subject (I.1) and of “toe-walking” in two younger subjects (III.4 and III.7). The possibility of either additional systemic manifestations of DOA hitherto unreported, or the presence of a contiguous gene syndrome owing to deletion of adjacent genes led us to investigate this family further. We learned of a brother (I.5) of the first generation with progressive leg weakness and hyporeflexia. Upon clinical evaluation, he was found to show no signs of optic atrophy. A niece of II.4 was reported to suffer from multiple abnormalities including syndactyly, asymmetry, growth retardation, and generalised muscle wasting (notably worse in the legs), but no optic atrophy. DNA was obtained from the brother (I.5), the niece (III.9), and her father (II.5). To determine whether or not these family members were carriers of the deletion, we performed genotype analysis. The results clearly show that none of these people is positive for the deletion. Therefore, we conclude that the other abnormalities are not segregating with DOA in this family and are not part of a contiguous gene syndrome.
Acknowledgments
The first two authors contributed equally to this work. The authors would like to thank the members of this family, without whom the study would not have been possible. The financial support of the Wellcome Trust (grants 035535/Z96 and 054529/Z98), the National Eye Research Centre, the Trustees of St James’s University Hospital, and the Ophthalmic Research Institute of Australia is also gratefully acknowledged. NJM is supported by the Emma and Leslie Reid Scholarship, University of Leeds.