Article Text

Statistics from Altmetric.com
SHOX (Short HOmeoboX containing gene) (OMIM 312865) is the single gene found in the “short stature critical region”, a 170 kb DNA segment of the pseudoautosomal (PAR1) region identified through genotype/phenotype correlations in X/Y abnormalities.1 The finding of a mutation generating a premature stop codon in exon 5 of SHOX, cosegregating with idiopathic short stature (ISS), provided evidence for the involvement of this gene in growth retardation, including the short stature of Turner syndrome.1 At the heterozygous level, large deletions or point mutations of the SHOX gene have been found in families affected by Leri-Weill dyschondrosteosis (LWD, OMIM 127300), a dominantly inherited skeletal dysplasia with disproportionate short stature owing to mesomelic shortening of the forearm and lower leg and Madelung deformity of the arm.2,3 In addition, the biallelic inactivation of the SHOX gene was shown in fetuses with Langer-type mesomelic dysplasia (OMIM 249700),2,3 a recessive form of dwarfism which was confirmed as the homozygous counterpart of LWD, as previously proposed on a clinical genetic basis.4
Some authors reported mutations in all the LWD cases studied,3,5 whereas others found SHOX mutations in about 60% of the cases.6,7 The same proportion of SHOX gene mutations are reported in the present study based on the analysis of a large group of Italian LWD families.
METHODS AND RESULTS
Patients and families were recruited, after informed consent, in the context of a collaborative study in several Italian paediatric endocrinology centres. Height and sitting height were measured to nearest 0.1 cm using a Harpenden anthropometer and measurements were converted into centiles, according to the Tanner tables. All subjects were submitted to x ray examination including the forearms and lower legs.
Inclusion criteria for the study were: (1) normal karyotype, with lack of demonstrable sex chromosome abnormality using standard methods; (2) disproportionate stature owing to mesomelic leg shortening, attributed when the upper/lower segment ratio was >1.1; (3) Madelung deformity of the forearms, attributed when all the following radiological features were present: bowing of the radius, triangularisation of the normally quadrangular shaped outline of the distal radial epiphysis with the apex of the triangle pointing medially, and dorsal dislocation of the ulna leading to a widened gap between the radius and ulna.8 For inclusion in the study, patients had to fulfil all three criteria.
A total of 42 patients from 21 families were studied, originating from northern Italy in 10 cases, from central Italy in four, and from southern Italy in seven. All probands (three males and 18 females) were of prepubertal or peripubertal age; three cases, all prepubertal females, were sporadic, whereas in the remaining cases there was a dominant pattern of inheritance.
DNA extracted from peripheral blood was used for PCR amplification reactions for the microsatellite sequences DXYS233, CASHOX, and DXYS234, in order to evaluate the presence of deletions in the PAR1 region encompassing the SHOX gene. The DXYS233 sequence is telomeric to the SHOX gene, CASHOX is an intragenic CA repeat localised in the 5` untranslated region of SHOX exon 1, and DXYS234 is located about 2 cM from the telomere.2,3 Loss of heterozygosity at loci DXYS233, CASHOX, and DXYS234 was used to establish the presence of deletions. For each of the 21 families under investigation, the proband, both parents, and other affected relatives (when present) were analysed through PCR amplification of the three fragments, using fluorescent dye labelled primers (conditions and primer sequences are available on request). Direct sequencing for all the coding exons of the SHOX gene (from exon 2 to exon 6a) was performed in all cases negative for deletion, using the ABI Prism BigDye Terminator Cycle Sequencing Ready Reaction Kit (PE Applied Biosystem) according to the protocol supplied by the manufacturer. Electrophoresis and analysis of microsatellites and sequences were run on a 377 DNA sequencer (PE Applied Biosystems).
Of the 21 probands meeting the clinical inclusion criteria for LWD, SHOX gene mutations were found in 13 (62% of the total). No significant difference was seen in the rate of mutation detection between familial (11 out of 18) and sporadic cases (two out of three cases).
In 10 families (48% of the total), a deletion encompassing the SHOX gene was found. In seven cases the deletion was interstitial: the CASHOX locus was selectively deleted in four families, whereas it was involved in association with the more centromeric marker (DXYS234) in three cases. In three families the deletion involved all the microsatellite markers. Deletions in both sporadic cases were interstitial.
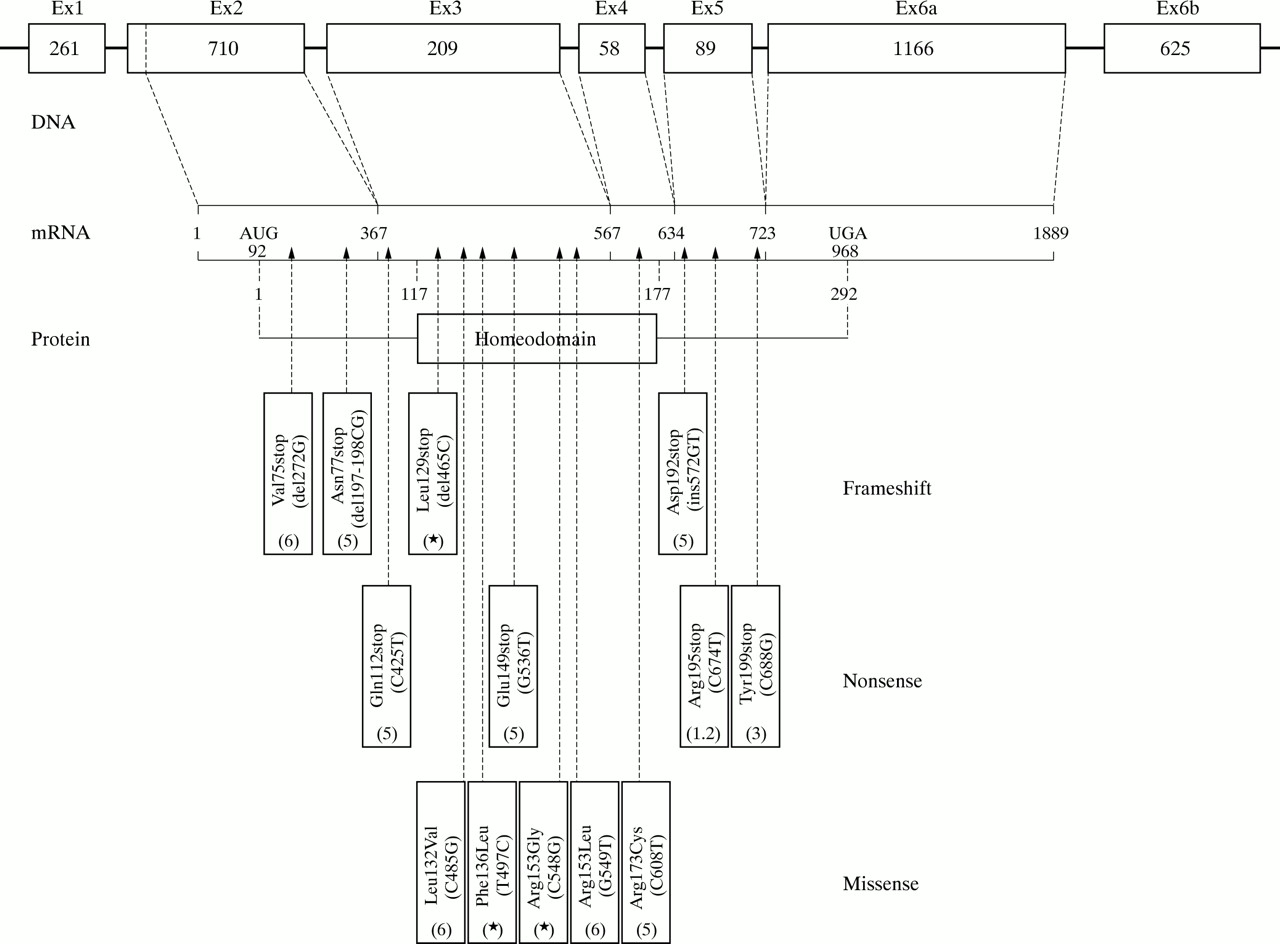
Out of the remaining 11 probands negative for a deletion, sequence analysis detected a SHOX gene mutation in three cases. All of them occurred in the homeodomain region: one was a frameshift (465delC) introducing a stop codon in the amino acid position 129, while the two others were missense mutations, both cosegregating with the disease in the family. In family 2 a T to C transition at nucleotide position 497 changed the highly conserved phenylalanine residue 136 to leucine, while in family 17 a C to G transversion at position 548 changed the arginine residue to glycine in the already reported altered amino acid residue 153 by Grigelioniene et al6 (fig 1). In eight cases (seven familial and one sporadic), no SHOX mutation was detected.

{kind=link}
Structure of the SHOX gene. The point mutations published so far and their position are indicated according to the GenBank sequence Y11536.
No differences in the clinical findings between families with or without SHOX mutations were seen. Madelung deformity and disproportion between the upper and lower segment were inclusion criteria and thus were present in all the patients (table 1), while height under the 3rd centile was present in 11 probands only (52%); seven of these patients (64% of the patients under the 3rd centile) harboured a SHOX mutation, whereas in four (36%) no evidence of mutation was found. Moreover, no genetic difference was seen in the families with SHOX mutations as compared to those testing negative for mutations.
Clinical features of the 21 LWD families and results of the mutation analysis
The results of the mutation analysis and the main clinical data of the LWD probands and their families are summarised in table 1.
DISCUSSION
In the present study, a wide sample of Italian families, representative of the whole country, were selected through probands affected by LWD. Idiopathic short stature, other forms of mesomelic short stature, and cases with isolated Madelung deformity were excluded. Although the criteria adopted (the “pure” LWD phenotype at prepubertal and peripubertal age both in males and females) were stringent, mutations involving the SHOX gene were found in 62% of the families only. Considering our data and all the available evidence from previous studies, at least a quarter of the families with the LWD phenotype do not have a mutation in the SHOX gene (table 2), with no phenotypic trait differentiating the “mutation positive” from the “mutation negative” families. Moreover, mutation positive and mutation negative families are also identical on a genetic basis, having a similar ratio of familial (always showing a dominant pedigree pattern) to sporadic cases of the syndrome.
Analysis of SHOX mutations in LWD families
In our series, SHOX gene mutations in LWD were point mutations in about a quarter of the probands (3/13) and deletions in the remaining cases (10/13), in accordance with the currently available data (table 2). Also in accordance with previous results7 is the finding that the majority of the deletions encompassing the SHOX gene were interstitial (7/10 cases). These peculiar findings of SHOX gene deletions (number exceeding the number of points mutations, and type mainly interstitial) are largely supported by the special structural features and the high recombination frequency of the pseudoautosomal PAR1 region.9 The three point mutations found in our families were all located in the homeodomain region of the SHOX gene; interestingly, the T497C transition coding for the Phe136Leu (fig 1) occurs in a highly conserved domain of the homeobox, similar to the missense mutation reported by Grigelioniene et al6 in another conserved region, while the other missense mutation alters a residue (Arg153Leu) which is less evolutionarily preserved, but is also mutated in another LWD patient.6 These residues do not appear to make direct contact with target DNA, but they are likely to be important for the stability of the homeodomain. The distribution along the SHOX gene of the 13 point mutations described so far in LWD shows that they are all private mutations, excluding one, which has also been found in an ISS case with mild rhizomelic body disproportion (fig 1).1 Missense mutations, causing semiconservative changes, are all concentrated in the homeobox region, while outside the region there is a prevalence of frameshift and nonsense mutations, indicating that a mutation outside the crucial region must necessarily truncate the protein to be functionally effective (fig 1). This finding gives some insight into the relationship between LWD, ISS, and SHOX gene mutations: in a large investigation carried out in 970 subjects with ISS, deletions and point mutations involving the SHOX gene were found in around 2-4% of the cases. Interestingly, however, the authors report mainly missense mutations, all sparing the homeobox region and possibly accounting for the milder phenotype of ISS compared to the LWD.10 Finally, the absence of correlation among the various degrees of deletion of the pseudoautosomal PAR1 region and the phenotypic involvement in LWD, as well as the lack of a specific clinical effect for differentiating LWD families with SHOX gene deletions from those having point mutations (either inside or outside the homeobox region), lend further support to the haploinsufficiency hypothesis as the mechanism underlying SHOX linked LWD.2,3,7
Key points
-
SHOX (Short HOmeoboX containing) gene mutations have been investigated in 21 unrelated families affected by Leri-Weill dyschondrosteosis (LWD), included in the study because of the presence of Madelung deformity of the forearms and disproportionate stature. Genomic deletions encompassing SHOX were assessed through the analysis of microsatellites within and flanking the gene. In non-deleted cases mutation analysis was performed through sequencing of the coding regions of the gene.
-
Ten out of the 21 families studied showed SHOX gene deletions. In the remaining 11 families, sequence analysis showed three novel point mutations (one frameshift and two missense).
-
Thirteen out of 21 (62%) families with typical LWD harbour a SHOX gene mutation; point mutations, predominantly involving the homeobox region, occur less frequently than large deletions.
In conclusion, from the available data we assume that SHOX gene mutations are the main cause of LWD, with no positive evidence yet existing of the involvement of other genes in the pathogenesis of the disease. Moreover, mutations affecting the level of SHOX expression can currently go undetected: in fact, interstitial deletions, already noted in the 3` end of the SHOX gene,3 mutations in the promoter or in the untranslated regions like exon 1 or exon 6b, or more distant mutations exerting position effect cannot be ruled out with the screening method adopted and might still possibly account for at least some of the mutation negative LWD cases.