Article Text

Statistics from Altmetric.com
- ATS, Alport syndrome
- MR, mental retardation
- ATS-MR, Alport syndrome and mental retardation
- ATS-DL, Alport syndrome and diffuse leiomyomatosis
- AMME, Alport syndrome, mental retardation, midface hypoplasia, and elliptocytosis
X linked Alport syndrome (ATS, OMIM 301050) is a hereditary glomerulonephritis resulting from either point mutations or intragenic deletions of the COL4A5 gene encoding the α5 chain of type IV collagen.1–3 Contiguous gene syndromes are phenotypically complex disorders associated with the deletion of multiple adjacent genes. There are several examples of such syndromes on the X chromosome.4, 5 Until recently, the only known contiguous gene syndrome involving the COL4A5 gene was Alport syndrome and diffuse leiomyomatosis (ATS-DL, OMIM 308940),6–9 in which the deletion extends towards the centromere to include the first two exons of the adjacent COL4A6 gene. In 1998, we described a new Xq22.3 contiguous gene syndrome which we named AMME (OMIM 300194) because of the distinctive features observed in affected males: Alport syndrome (A), mental retardation (M), midface hypoplasia (M), and elliptocytosis (E).10 After the original publication, clinical re-evaluation of the family showed alterations of cardiac rhythm and morphology on echocardiography.11 To elucidate the molecular basis of this complex syndrome, we cloned three genes from the deleted region: FACL4,12AMMECR1,13 and KCNE1L,11 recently renamed KCNE5.14 Now we report the characterisation of the deletion, which extends for about 2 Mb, and the identification of a second family which bears a smaller deletion of about 1 Mb. Comparison with two other deletions extending beyond COL4A5 in the telomeric direction and generating only ATS allowed us to define the critical region for mental retardation (MR), which contains four candidate genes. We now propose renaming this condition ATS-MR, so that consistent terminology is used to describe the known ATS contiguous gene syndromes.
METHODS
FISH (fluorescence in situ hybridisation) analysis was performed essentially as described by Lichter et al15 with minor modifications.
Somatic hybrid construction from a lymphoblastoid cell line of the carrier mother of family 2 was performed as previously described.16 Selection for the active X chromosome yielded 12 clones bearing only the normal X and five clones retaining both X chromosomes. Back selection for the inactive X on one diploid clone yielded 12 hybrids containing the deleted inactive X chromosome, three of which were selected for analysis. Chromosome painting, performed on metaphase chromosomes preparations with an X chromosome specific probe (P5222-DG.5, Oncor),17 confirmed that these hybrids contained only one X chromosome.
A long range deletion map was constructed using available X chromosome YAC contigs and the DNA of the proband of family 1, hybrid 1C3 derived from the mother of family 2, and patients 3 and 4. The extent of these four deletions was compared with a previously reported deletion present in patient 5, diagnosed with ATS-DL.6, 9 The partial X chromosome maps published in 199518 and those available at the Max-Plank-Institut für Molekulare Genetik in Berlin and at Massachusetts Institute of Technology (MIT: http://www-genome.wi.mit.edu/ftp/distribution/human_STS_releases/oct96/pictures/ChrX.hqx) have been used to obtain an STS series. Marker D4-2 has been drawn inside PAC clone RP3-465D4 (GenBank accession no AL360174) and primer sequences are the following: D4-2S: 5` tcagctacaatctgcctaag 3`; D4-2A: 5` ctcttgggaccaagcagtc 3`.
Southern blot analysis was performed with the following probes: for PPP6C we used an EcoRI/NotI/PstI fragment obtained from IMAGE clone 782102 (GenBank accession No AA431365); for GUCY2F and NXT2 we used two markers drawn on the ends of PAC RP4-596C15 (GenBank accession No AL0313687), which contains both genes (primers: C15F1: 5` actgacctacatagctgactg 3`; C15R1: 5` ataaacaatggcatcttaggag 3`; C15F3: 5` ctgacatgcagatgcatgttg 3`; C15R3: 5` agctcagttcaccttcattatg 3`); for TDGF3 the probe CR-1-P719 was used; for IRS4 primers were designed at the 5` end of the gene (IRS-F: 5` aggctgtggtcacgtgttg 3`; IRS-R: 5` accatcgcgaagtattcgtc 3`). A panel of YACs from various sources (CEPH, ICI, and YCRF libraries) was tested with the above markers.
X inactivation analysis was performed as described by Allen et al,20 with the modifications reported in Meloni et al.21
PATIENTS
We have previously reported that family 1 has a COL4A5 gene deletion10 extending beyond the gene towards the telomere12 (AMME, OMIM 300194). In order to find additional patients with a deletion in Xq22.3, we carefully examined previous publications and found two interesting reports in which nephropathy was associated with deafness and mental retardation. One was written in 1962 and the patients also had hyperprolinaemia,22 while the second, written in 1994, showed that the patients also had macrocephaly.23 Only one sample from the obligate carrier mother of the second reported family (family 2) was available, since the male patients had died.
We also characterised two additional patients (3 and 4) with isolated ATS caused by a deletion extending beyond COL4A5 towards the telomere. These deletions were compared to that found in a fifth patient diagnosed with ATS-DL contiguous gene syndrome, whose deletion extends beyond COL4A5 towards the centromere.
ATS-MR patients
Family 1
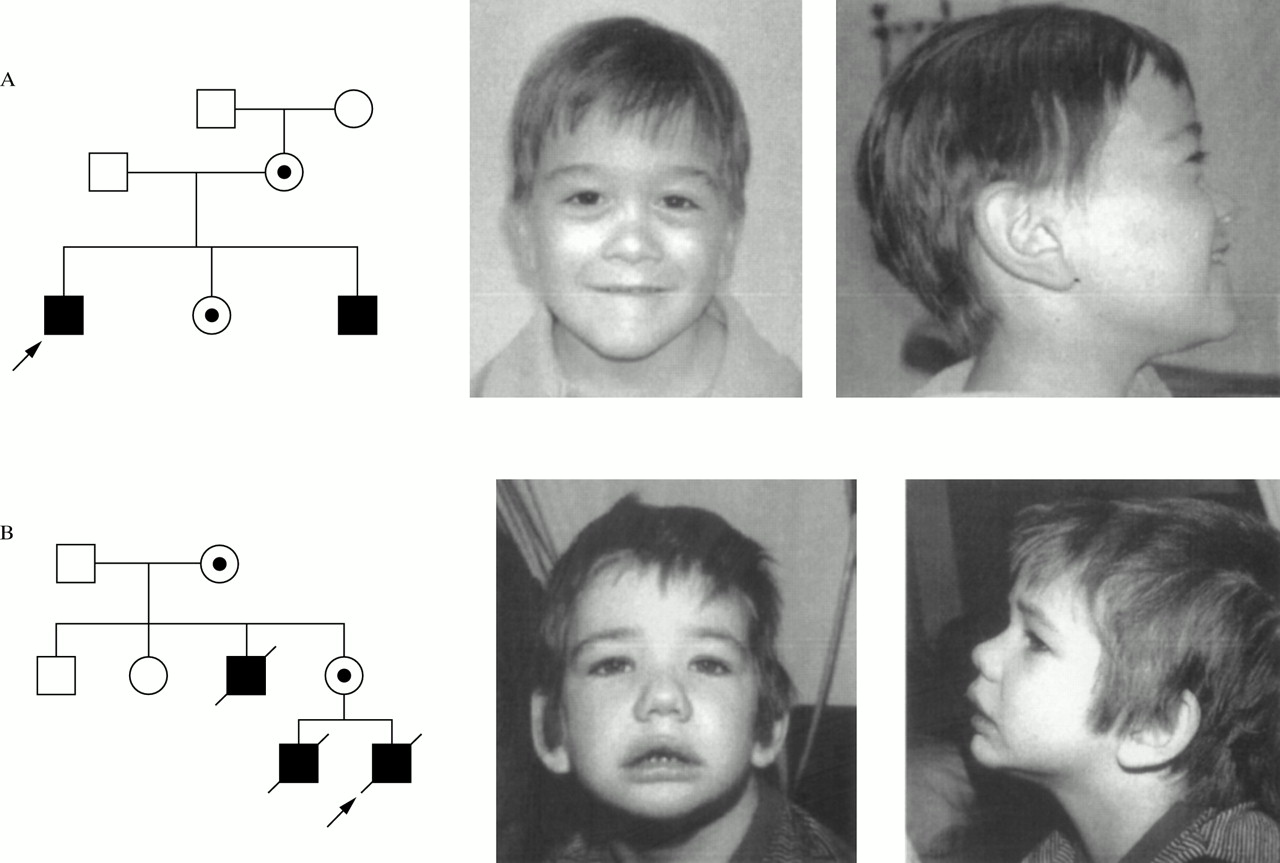
The family of proband 850 (indicated by an arrow in fig 1A) is composed of four relevant members, a mother, two sons, and a daughter, who all have microhaematuria and, except for the daughter, hypoacusia. The two males have additional features including MR, dysmorphic facies with marked midface hypoplasia, and elliptocytosis.10, 11 Follow up of patients after the original observation shows that they remain in good medical health. The proband has significant myopia, stable bilateral hearing loss, and haematuria with normal renal function and blood pressure. His current EKG shows right bundle branch block and normal sinus rhythm. His echocardiogram shows a small PDA, a non-stenotic bicuspid aortic valve, and mild left ventricle dilatation. Pertinent physical findings include 50th centile head circumference, mild midface hypoplasia, and three supernumerary teeth; the remainder of his examination is unchanged from that in the previous report. Developmentally, at the age of 12 years he is functioning at a 4 year level. His brother has astigmatism and microstrabismus, bilateral mild to moderate hearing loss, microscopic haematuria with normal renal function, and developmental delay. His head circumference is on the 25th centile and physical findings are unchanged from the previous report. His EKG is normal at present while his echocardiogram shows mild mitral and tricuspid regurgitation. The mother and sister of the boys remain healthy. Investigation of the mother for leiomyomas has been negative.

Pedigree and facial appearance of probands of families 1 (A) and 2 (B). Probands are indicated by an arrow.
Family 2
The first clinical description of this family (fig 1B) was reported by Robson et al.23 The family's relevant members are a grandmother, an uncle, a mother, and her two sons (the proband and his older brother). The three males presented with a hereditary glomerulonephritis while the two females had microhaematuria. All three males also had MR and both the proband and his brother had macrocephaly; moreover, the proband also had metaphyseal dysostosis. Re-evaluation of the patients' clinical data showed no evidence of elliptocytosis in the proband or his older brother. EKG findings for the proband were normal on more than one recording. The macrocephaly described in both the proband and his brother was also present in their normal father but not in the affected uncle. Additional information about the three affected males was not available since they have all died.
Comparison of dysmorphic features
Based on photographic review (fig 1), similar dysmorphic features in the boys of families 1 and 2 include anteverted nares, flat nasal bridge, mild midface hypoplasia (far more pronounced in family 1), downward slanting palpebral fissures and peripalpebral fullness (more pronounced in family 2), facial hypotonia, hair whorls, and fetal fingertip pads (not shown). Moreover, the children of both families become less dysmorphic as they get older. The boys are discordant for the presence of full lips in family 2 and thin lips in family 1. Since the macrocephaly described in the proband of family 2 and his brother is also present in their normal father, it is likely to be a familial finding not related to the syndrome. There is clearly phenotypic similarity but not identity between the male probands in each family.
ATS patients
Patient 3
A detailed clinical description of this patient, previously identified as patient MC, has been reported elsewhere.24, 25 The patient had isolated ATS with persistent proteinuria and haematuria, sensorineural hearing loss, and typical renal biopsy features and progressed to end stage renal failure at the age of 19.
Patient 4
Patient 4 is a 22 year old male, who presented with haematuria and proteinuria since the ages of 9 and 12, respectively. Moderate hypoacusis in all frequencies was diagnosed at the age of 12. Ocular examination was normal. He progressed to end stage renal failure at the age of 17. He has a younger brother with a very similar phenotype.
ATS-DL patient
Patient 5
Patient 5, previously described as patient DON,6, 9 is a 30 year old man, diagnosed with ATS at 6 years of age. He progressed to end stage renal failure by the age of 16 and subsequently underwent renal transplantation. Oesophageal leiomyomas were diagnosed during his early twenties.
RESULTS
FISH analysis on the carrier mother of family 2 showed monosomy for YACs (ICI-9DA12, ICI-9GF9) covering the Xq22.3 region (not shown). This confirms the presence of a Xq22.3 deletion in family 2. In order to define the size of the deletions in both families 1 and 2 we performed additional FISH experiments. In family 2, the two breakpoints were positioned inside clones yCOLPF-1 and 900G0974 (fig 3A). In family 1, the deletion was larger. YACs 813b10 and 896d2 gave an overlapping signal on one X chromosome of the carrier mother when used together (fig 2), defining the proximal and the distal breakpoints, respectively, in this family (fig 3A).
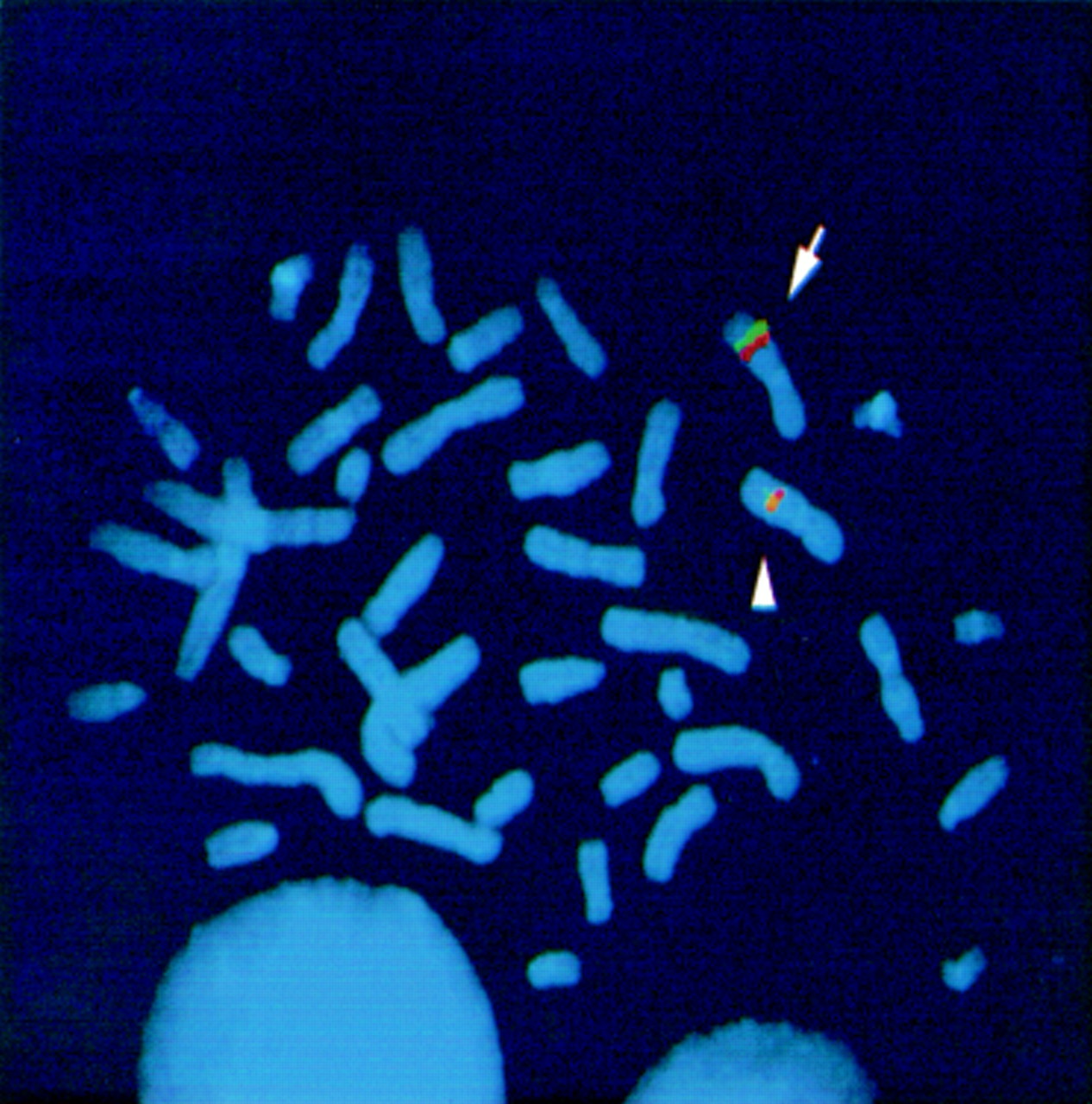
FISH analysis. Metaphase chromosome preparation from the carrier mother of family 1 hybridised with YACs 813b10 (green) and 896d2 (red). The two clones give separate signals on the normal X chromosome (arrow) while they give an overlapping signal on the deleted chromosome (arrowhead).

Physical map. (A) Genes (italics) and markers (plain text) located in the region of interest are indicated at the top. SGC8439 and WI-8307 (italics) represent two ESTs. Extensions of deletions in the two ATS-MR (1 and 2), the two ATS (3 and 4), and the ATS-DL patient (5) are shown as shaded bars. Minimal critical region for MR is also indicated. YACs covering the region are reported at the bottom. An asterisk indicates YACs used in FISH analysis. Clones yCOL1-1, 9DA12, and 9GF9 were all deleted from one X chromosome of the carrier mother of family 2. In contrast, 900G0974 and 19CG9 were present on both chromosomes. Towards the centromere, yCOLPF-1 was not deleted. In the mother of family 1, the large YAC 744a12 was entirely deleted from one X chromosome. Conversely, the telomeric clones 896d2 and 954d9 were present on both chromosomes with the same signal intensity. Towards the centromere, 813b10 was present. YACs 813b10, yCOL1-1, yCOLPF-1, 851d2, 656g10, 744a12, 896d2, 749f7, 737h4, and 954d9 are from CEPH. YACs 33BB12, 9DA12, 9GF9, 19CG9, 124G1, and E110H9 are from ICI, and YACs 900G1227, 900F0676, 900G0974, and 900E8148 are from YCRF. (B) Magnification of MR minimal critical region delimited by the distal deletion breakpoints of patient 3 and of family 2 showing the genes included (top). Arrows below each gene name indicate the direction of transcription. The region also contains a partial sequence highly homologous to PPP6C, located between KCNE5 and the 3` half of FACL4 (not shown). Two representative YACs covering the region are indicated as shaded lines. At the bottom, sequenced clones covering the region derived from the Sanger Centre (clones RP1-31B8, RP3-596C15, RP1-136J15, RP6-13I1 and RP1-205E24) and from Celera Genomics public database (scaffolds x2HTBKY6TRW and x2HTBKUMC41) are drawn. Black dots indicate that the exon or the marker is contained in the clone.
STS analysis on family 1 showed that the deletion extends from the second intron of the COL4A6 gene to marker D4-2. The distal breakpoint is located in a region defined proximally by marker 954D9L (deleted) and distally by marker D4-2 (present) (fig 3A). The proximal breakpoint is located between exon 3 of COL4A6 (present) and marker E7M13 (absent) (fig 3A). Based on the YAC contig, the deletion has an extent of about 2 Mb.
PCR and Southern blot analysis on hybrid 1C3 (derived from the mother of family 2) showed that the deletion begins in intron 47 of the COL4A5 gene and extends towards the telomere partially to include the gene FACL4, with the distal breakpoint located in intron 2 of the gene (fig 3A, B). Based on the dimension of YACs covering the region, this deletion has an extent of about 1 Mb.
Ding et al25 identified a COL4A5 gene deletion in patient 3 and located the proximal breakpoint near intron 3. Further analysis allowed us to show that the deletion extends beyond the gene towards the telomere, with the distal breakpoint located between EST SGC8439 (deleted) and marker DXS1210 (present) (fig 3A, B). Patient 4 has a smaller deletion which starts in intron 18 of the COL4A5 gene and extends beyond exon 51 up to marker 2B6, which is located 2 kb from the end of COL4A5.
Comparison of the four deletions described above allowed us to define a deletion map of the region (fig 3A) and to delimit a minimal critical region containing candidate genes for MR. This MR critical region is included between the distal breakpoints of patient 3 and family 2 and has an extension of about 380 kb (fig 3B). It is completely covered by four sequenced PACs from the Sanger Centre (http://www.ensembl.org/) and by scaffold x2HTBKY6TRW from the Celera Genomics public database (http://publication.celera.com/)26 (fig 3B). Four genes are confined in this MR minimal critical region, deleted in both ATS-MR families but not in ATS patients. They are, from centromere to telomere, GUCY2F, NXT2, KCNE5, and FACL4 (fig 3A, B). The minimal critical region also contains a partial sequence highly homologous to the serine/threonine phosphatase PPP6C (GenBank accession No X92972).27
All deletions analysed encompass the gene COL4A5. The deletion present in family 1 extends into the first two exons of COL4A6. PCR analysis has shown that the IRS4 gene is deleted in both families 1 and 2, as well as in patient 3. The gene AMMECR1, deleted in family 1, is intact in family 2. In fact the deletion of family 1 extends a large way towards the telomere and also includes GNG5L and TDGF3.
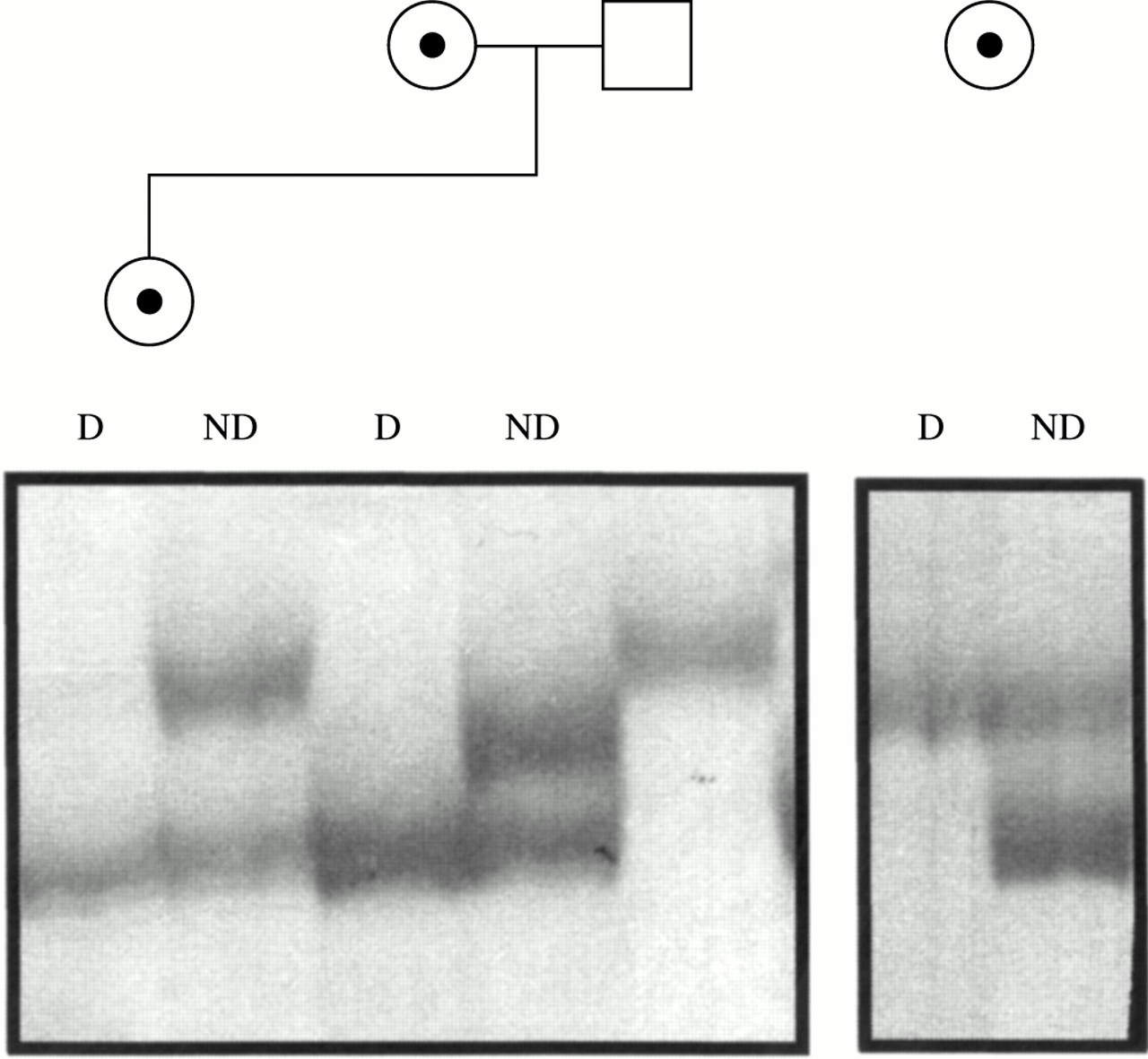
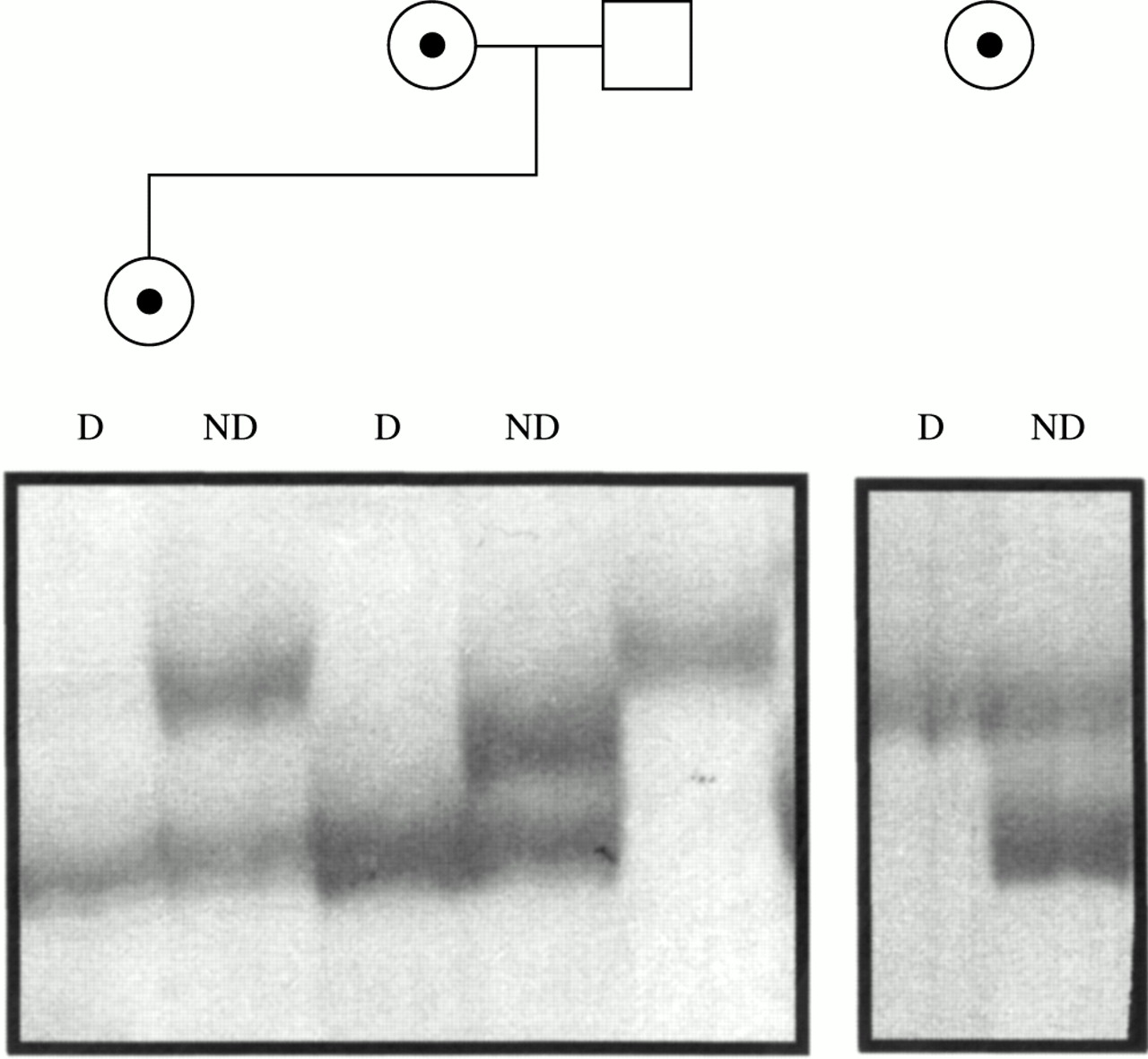
X inactivation analysis, performed on the carrier mother and sister of family 1 (fig 1A) and on the carrier mother of family 2 (fig 1B), showed a completely skewed pattern in all three females (fig 4). Segregation analysis in family 1 showed that, in the sister, the inactive X chromosome is the maternal one carrying the deletion. A completely skewed X inactivation in the mother of family 2 is in agreement with the results of somatic hybrids. All hybrids (12 out of 12) selected for the active X chromosome carried the normal X, while all clones obtained by back selection for the inactive X yielded the deleted X chromosome, suggesting that in this female also the skewed X inactivation is in favour of the normal X.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
X inactivation assay. Six percent polyacrylamide silver stained gel of PCR products of the CAG repeat of the androgen receptor gene from HpaII digested (D) and non-digested (ND) DNA of the two carrier females of family 1 (left panel) and of the carrier mother of family 2 (right panel). For family 1, the allele present in the father is also shown.
DISCUSSION
Contiguous gene syndromes are phenotypically complex disorders produced by the deletion of multiple adjacent genes. The discovery of patients with contiguous gene syndromes has facilitated the cloning of genes responsible for monogenic diseases. Ballabio et al5 studied a complex patient with a large deletion on the tip of Xp which was thereafter shown to be the synergy of at least six monogenic diseases. The identification of single genes was accomplished by the construction of a deletion map using several patients with different deletion sizes. The same strategy was applied in the present study for the ATS-MR contiguous gene syndrome on Xq22.3.
Our ATS-MR patients have a deletion that encompasses the COL4A5 gene, responsible for the Alport phenotype.1 Besides Alport syndrome, the most consistent feature observed in this contiguous gene syndrome is mental retardation. Up to now, nine genes have been associated with non-specific X linked mental retardation28: RSK2 on Xp22.2-p22.1, IL1RAPL1 on Xp22.1-Xp21.3, TM4SF2 (also known as MSX1) on Xp11.4, OPHN1 on Xq12, PAK3 on Xq21.3-q24, ARHGEF on Xq26, and GDI1, FMR2, and MECP2 on Xq28. Two of these genes (OPHN1 and IL1RAPL1) are also involved in X linked contiguous gene syndromes.29, 30 Two possible candidate genes for MR in ATS-MR syndrome could be IL1RAPL2, which maps to Xq22 and shares 65% identity with IL1RAPL1,30, 31 and PAK3. Although a position effect on either of the two genes cannot be definitively excluded, neither of them is deleted in our two ATS-MR families.
In this paper, we confirm the presence of a mental retardation contiguous gene syndrome at Xq22.3. Several MRX families are mapped to a region that includes Xq22.3. Among these, MRX3032 and MRX4733 have been associated with mutations in PAK3, a serine/threonine kinase involved in the Rho GTPase signalling and highly expressed in the cerebral cortex and hippocampus.32 For other linked families, such as MRX23,34 MRX35,35 MRX53,36 and MRX68 (XLMR Genes Update Web Site: http://xlmr.interfree.it/XLMR/Tab5.html), the responsible genes remain to be elucidated. We suggest that one of the four genes included in our MR minimal critical region (FACL4, KCNE5, NXT2, and GUCY2F) may be the causal MR gene for at least one of the above MRX families. At present, the most interesting candidates are FACL4, which is an acyl-CoA synthetase with high substrate preference for arachidonic acid,37 highly expressed in the developing and adult brain,38 and NXT2, which is important for efficient mRNA export from the nucleus.39 However, KCNE5 also seems a promising gene because of its restricted pattern of expression in CNS, heart, and skeletal muscle.11 Involvement of GUCY2F in the causation of MR seems unlikely, considering that its expression is restricted to a particular subtype of retinal cells, at least in mouse and rhesus monkey.40, 41
A distinct dysmorphic phenotype is associated with MR in our two families. At present, it is difficult to know whether the dysmorphic features are the result of the deletion of a gene important for facial development or if they are a pleiotropic effect of a MR gene. The latter hypothesis is supported by reports that the same gene can be responsible for both specific and non-specific MR. RSK2, a growth factor regulated serine/threonine kinase, has proven to be responsible for Coffin-Lowry syndrome42 and is also mutated in the non-specific MRX19 family.43 More recently, we and others reported that the gene silencer, MECP2, responsible for Rett syndrome, may be involved in specific21 and non-specific MR.44 Some specific MR syndromes map to Xq22. One family with moderate mental retardation, congenital hip dislocation, microcephaly, and dysmorphic facial features was reported by Carpenter et al.45 An X linked MR syndrome with seizures, hypogammaglobulinaemia, and progressive gait disturbance was mapped between Xq21.33 and Xq23,46 and another with short stature was mapped in Xq24.47 One of the genes included in our MR minimal critical region may be contributing to the mental impairment in these families.
-
Identification of a second family with Alport syndrome and mental retardation.
-
Confirmation of a contiguous gene syndrome in Xq22.3 which we propose to call ATS-MR.
-
Characterisation of the deletions in the two ATS-MR families.
-
Definition of a minimal critical region for mental retardation containing four candidate genes, FACL4, NXT2, KCNE5, and GUCY2F.
The deletion in family 1 extends beyond the MR minimal critical region both towards the centromere and the telomere. Towards the centromere, the deletion includes the IRS4 gene,48 located 34 937 bp downstream of COL4A5. IRS4 is deleted in both families 1 and 2 and also in patient 3, an adult with isolated ATS; therefore this gene is excluded from the MR minimal critical region, as it appears to have no phenotypic effect. Towards the telomere, the large deletion of family 1 includes three additional genes, AMMECR1 and GNG5L, which could be a processed pseudogene, and TDGF3, a possibly transcribed pseudogene with high sequence identity to teratocarcinoma derived growth factor 1 (TDGF1).19 Since this region is not completely sequenced, we cannot exclude the presence of additional genes. The contribution of any of these genes or pseudogenes to the complex phenotype of family 1 is unknown.
Comparison of the five deletions described in this paper allowed us to define a deletion map of the region. This deletion map confirmed the marker order reported by the Celera Genomics public database and by the Sanger Centre, except for a segment included between markers DXS456 and SGC8439. According to both Celera and Sanger Centre sequences, the order of genes and markers in this region should be cen-DXS456-FACL4-KCNE5-NXT2-GUCY2F-DXS1210-SGC8439-tel. This order is not compatible with our deletion map unless we hypothesise complex deletions in both family 2 and patient 3. We conclude that the correct order is cen-SGC8439-DXS1210-GUCY2F-NXT2-KCNE5-FACL4-DXS456-tel (fig 3A). In fact, the present order of genes and markers in the updated private Celera database is now corrected and conforms to our proposed order26 (M Adams, personal communication).
In conclusion, we have further characterised the new contiguous gene syndrome in Xq22.3, which we propose to call ATS-MR; this adds to the previously known contiguous gene syndrome in this region, ATS-DL (Alport syndrome and diffuse leiomyomatosis). Both syndromes involve deletion of COL4A5, but while ATS-DL extends centromerically, ATS-MR extends telomerically with respect to the collagen gene. Owing to the relative simplicity of identification of the second family, with reference to published reports alone, it can be hypothesised that this syndrome may be less rare than initially thought. However, search for additional patients with the ATS-MR contiguous gene syndrome will be necessary in order to verify the real frequency of this condition. Moreover, we have defined a critical region for MR, containing four candidate genes, FACL4, KCNE5, NXT2, and GUCY2F. Extensive screening for point mutations in these genes in patients with MR will allow the identification of the true gene responsible for MR in this disorder.
Acknowledgments
This work was supported by a Telethon Grant to AR (E1145) and by a “Progetto Giovani Ricercatori” (University of Siena) to FV. We thank Celera Genomics.