Article Text

Statistics from Altmetric.com
The common clinical features reported in ring chromosome 22 cases include overall developmental delay with severe speech disability, growth retardation with frequently associated microcephaly, hypotonia, and dysmorphic traits, such as epicanthus, normally placed but large and dysplastic ears, long eyelashes with full eyebrows, and occasionally high arched palate, dental malocclusion, and mild hypertelorism.1–9 Second and third toe syndactyly, unsteady gait, hyperactivity, aggressive behaviour, autistic disorders,3, 4 and seizures or abnormal EEG1, 2 have also been reported. The recently described subtelomeric 22q deletion syndrome showed overlapping clinical features with a generalised developmental delay, particularly severe in the area of expressive speech.10–14 The phenotype is usually more severe in cases of ring chromosome 22 compared with subtelomeric 22q deletions. The phenotypic differences, particularly the growth retardation with microcephaly and the severe mental delay, could be the result of a larger deletion size in ring chromosome 22 cases. We report the molecular cytogenetic analysis of a ring chromosome 22 in a young boy investigated for global development and severe speech delay. This is the first molecular analysis of a ring chromosome 22 harbouring the second smallest 22qter microdeletion reported so far. Characteristics of cases with ring chromosome 22 and subtelomeric 22q deletion are reviewed.
PATIENT, MATERIALS, AND METHODS
Case report
The patient, a 7 year old boy, was seen for evaluation of global developmental delay. He was born to healthy, non-consanguineous parents aged 35 (mother) and 39 (father) years. His two older sisters were normal. Pregnancy, delivery, and the neonatal period were uneventful. His birth weight was 3330 g (50th centile), length was 50 cm (25th centile), and head circumference was 35 cm (50th centile). At the age of 4 months, his weight, height, and head circumference were 6500 g, 62 cm, and 42.5 cm, respectively (50th centile). At the age of 20 months, his head circumference was 50.5 cm (>75th centile). Mild motor developmental delay was noticed from the 12th month; the child began to walk at 17 months. Evaluation at the age of 7 showed a generalised developmental delay with a patent discrepancy between development of speech and of the other areas. Difficulties in running and going down the stairs persisted, he could not ride a bicycle, and his manual ability was poor, but the clinical examination did not find any definite neurological syndrome. The language impairment was severe; he spoke his first words at the age of 6 and he constructed only simple sentences with difficulty at the time of examination. Behavioural problems were also noted, such as hyperactivity, short attention span, and aggressive outbursts. The only minor dysmorphic findings were a broad nasal bridge, long lashes, dental malocclusion, and a high arched palate (fig 1). Audiometry and EEG were normal.

Face of patient. Note broad nasal bridge and long eyelashes.
Cytogenetic analysis
Standard and high resolution R and G banding chromosomal analyses were performed on metaphase preparations obtained from peripheral blood lymphocytes of the proband and his parents.
Fluorescence in situ hybridisation (FISH)
Five fluorescent 22q13 specific probes were used: two commercially available probes, D22S39 (Appligene Oncor Inc, Illkirch) and D22S1726 (AmpliTech Cytocell Inc, Compiégne) and three BAC recombinants containing the markers D22S45, D22S55, and ARSA.
Metaphase spreads were prepared from peripheral blood lymphocyte cultures of the proband and his parents. Commercial probes were hybridised according to the manufacturer's instructions. BACs were biotinylated by nick translation (Roche Inc, Mannheim) and subsequently hybridised overnight after preannealing with Cot-1 DNA (Vysis, Inc) using standard protocols. For these probes, signal amplification was performed using fluorescein labelled avidin and anti-avidin antibodies (Oncor). Chromosomes were counterstained with propidium iodide (Sigma) or 4`,6-diamidino-2-phenylindole (DAPI) (Sigma). Preparations were examined using a Zeiss Axioplan 2 epifluorescence microscope with a triple band pass filter.
RESULTS
Cytogenetic analysis
Examination of 50 metaphases from the proband showed, in all cells, 46 chromosomes with a ring chromosome 22. His karyotype was designated 46,XY,r(22). Parental karyotypes were normal.
Molecular cytogenetic study
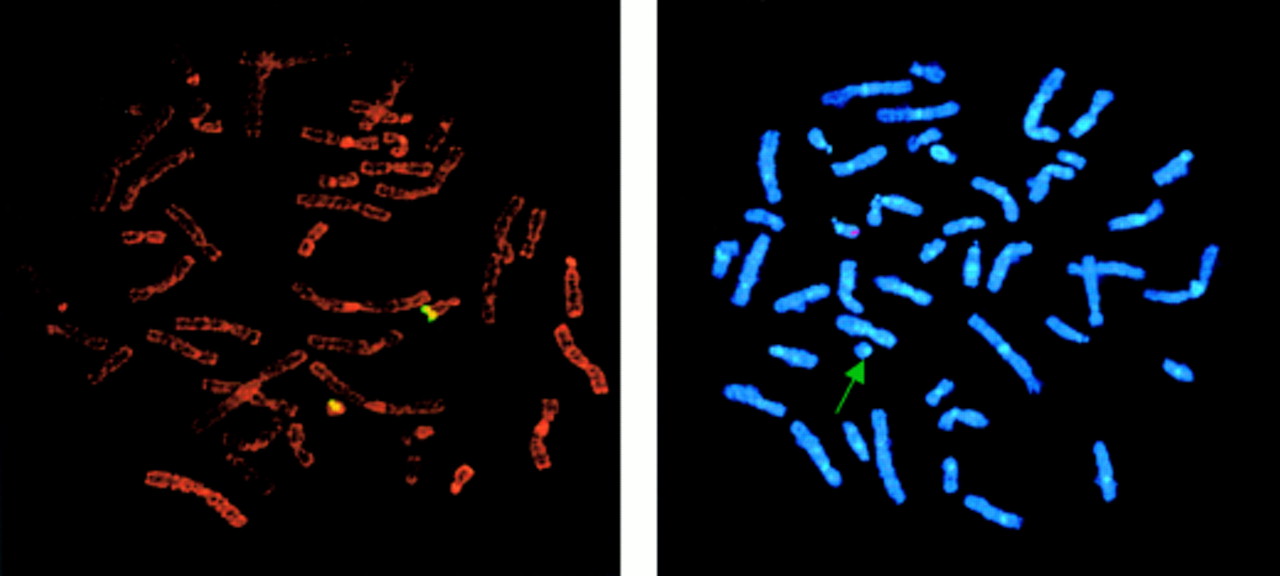
To characterise a putative 22qter deletion in the proband, we performed FISH experiments using a set of markers specific for five 22q13 loci (table 1). A hybridisation signal was present on both chromosomes 22 with the D22S45, D22S39, and D22S55 probes. On the other hand, the signal was absent from the ring chromosome 22 with the D22S1726 and ARSA probes and observed only on its normal homologue (fig 2).
Molecular data from simple subtelomeric 22q deletion studies and evaluation of the deleted segment size (bold). Loci are listed in order from most proximal to most distal15, 16, 20 (STS map on www.ncbi.nih.gov, September 2001)
{kind=link}
{kind=link}
FISH analysis of the proband. (Left) D22S55 fluorescence signals on normal and ring chromosome 22. (Right) D22S1726 fluorescence signal on only normal chromosome 22 (arrow, ring chromosome 22).
These results were highly suggestive of a small 22q deletion distal to D22S55 on the ring chromosome 22.
DISCUSSION
Severe speech delay is one of the main clinical feature observed in cases of ring chromosome 22 and 22q13 cytogenetically apparent and cryptic deletions.1, 10–16 Cytogenetic anomalies involving the terminal 22q arm may be associated with a severe phenotype, especially in the case of ring chromosome 22, where it includes profound mental delay, growth retardation, and microcephaly. On the other hand, most 22qter cryptic deletions show a less severe phenotype with milder mental retardation and few dysmorphic traits, while the severe speech impairment remains constant. Among the 10 22qter cryptic deletions reported so far,10–14, 17–19 the eight with clinical features described10–14 show the constancy of the language impairment. The other clinical features, dysmorphic traits, anomalies of the extremities, and autistic and behavioural disorders, are inconstant and shared by both ring chromosome 22 and 22qter deletions. The more severe phenotype in cases of ring chromosome 22 could be because of a larger deletion size. Hence, it is worth noting that in our proband mild mental retardation and normal growth and head circumference are associated with a cryptic 22qter deletion. It is tempting to assign this relatively mild phenotype to the small size of the deletion, which extends from D22S55 to the telomere and encompasses D22S1726 and ARSA. To date, 14 apparently simple 22q13 deletions have been studied.10, 13, 15, 16 All except one were larger than the present one (table 1). The smallest described so far10 had a breakpoint distal to ARSA corresponding to the 130 kb terminal 22q sequence.20 It is significant that the two smallest subtelomeric 22q deletions display a similar phenotype characterised by severe speech delay with absent or few dysmorphic features and no anomalies of the extremities. Therefore, our data indicate a phenotype-genotype correlation in the ring chromosome 22 syndrome and prompt us to consider the distinctive phenotypes of the ring chromosome 22 and the simple 22qter deletion as a consequence of contiguous gene dysfunction. More severe phenotypes in some 22q13 deletion cases could be the result of partial trisomies. In fact, recent data indicated that some subtelomeric deletions could be derived from unbalanced inherited cryptic translocations. An inherited chromosomal rearrangement was found in 10 out of 22 mentally deficient children harbouring a subtle chromosomal abnormality detected by a multiple subtelomeric probe assay.17 The association of cryptic 22q deletions with partial trisomies was specifically reported by Praphanphoj et al.19 These cases raise the question of an association of apparent simple 22qter microdeletions with undetected partial trisomies. It is noteworthy that the patient reported by Doheny et al18 was primarily considered as bearing a simple 22q deletion which was finally found to be associated with a partial 6p trisomy.19 These findings could explain the phenotypic differences in 22qter deletions and emphasise the interest of this observation in the characterisation of the pure subtelomeric 22q deletion phenotype and in the refinement of the critical region involved in language development. It is important to undertake a 22qter FISH analysis in patients with severe speech delay as the number of reports of 22qter cryptic deletion cases indicates that it may be frequently involved.
Acknowledgments
The first two authors contributed equally to this work. The authors are grateful to Mrs Kristina Schröder for providing the BAC recombinants.