Article Text

Statistics from Altmetric.com
- PPAR, peroxisome proliferator activated receptor
- PPRE, PPAR response elements
- BMI, body mass index
- HDL, high density lipoprotein
- LDL, low density lipoprotein
The brachydactylies are a group of inherited disorders characterised by shortened or malformed digits that are thought to be the result of abnormal growth of the phalanges and/or metacarpals. First classified by Bell into types A, B, C, D, and E, they were reclassified by Temtamy and McKusick1 and Fitch.2 Brachydactyly type A1 (BDA1, MIM 112500) is characterised by shortened or absent middle phalanges. Often the second and fifth digits, as well as the first proximal phalanx, are the most severely affected. In addition, all of the small tubular bones tend to be reduced in size and the metacarpals may be shortened, particularly the fifth metacarpal. Radial/ulnar clinodactyly, as well as malformed or absent epiphyses, have also been reported.1,2 Complex syndromes have been described in which BDA1 is one of a number of manifestations.3
Recently, genetic linkage for a BDA1 locus has been reported to map to 2q35-q36 in two unrelated Chinese families.4 Subsequent sequence analysis identified mutations in the Indian Hedgehog gene (Ihh) in affected subjects.5 There have been no other reports of linkage for BDA1, although identification of a balanced translocation between 5q11.2 and 17q23 in a girl with Klippel-Feil anomaly and BDA1 suggests that there may be a BDA1 locus on either chromosome 5 or 17. Mastrobattista et al6 examined a number of candidate genes, including HoxD, Msx1, Msx2, FGF1, and FGF2, in two families with BDA1, but did not find evidence of linkage. Genes involved in two of the other types of brachydactyly have been described. Mutations in CDMP1, a member of the TGF-β superfamily, have been found in a variant of autosomal dominant brachydactyly type C in which the middle phalanges of the second, third, and fifth fingers are shortened.7 Mutations in this gene also cause Hunter-Thompson and Grebe acromesomelic dysplasias, two autosomal recessive conditions.8 In addition, dominant mutations in ROR2, an orphan receptor tyrosine kinase, have been shown to cause brachydactyly type B.9,10
We have recently described a three generation family with mild BDA1,3 in which 13 affected subjects exhibited shortened middle and distal phalanges, proximal first phalanx, and fifth metacarpal. Consistently, the middle phalanges of affected members were below 2 SD of age matched norms. Most of the proximal first phalanges and fifth metacarpals of affected subjects were similarly 2 SD below the norms. Affected members also tended to be of short stature. The children who were studied had coned and prematurely fused epiphyses. Several members, such as the proband, had clinodactyly of one or more digits. A number of subjects also had a broad distal hallux and/or broad, slightly adducted forefoot. Since our initial report, we have ascertained an additional 15 family members including nine affected subjects. The phenotype of these additional family members is similar to those subjects in the family already described, and no additional clinical findings were associated with BDA1 in this family. We now report linkage of BDA1 in this kindred to a novel locus on chromosome 5.
METHODS
The linkage study comprised 34 members including 20 affected subjects and was conducted after approval by the Children's Hospital of Eastern Ontario Ethics Review Committee.
Peripheral blood samples were taken with informed consent from all participating family members, and a standard protocol was used to isolate DNA. A genome wide scan was initiated using 36 primer sets from the MAPPAIRS™ microsatellite markers (Research Genetics, Huntsville, Alabama), encompassing markers from 16 chromosomes. Particular emphasis was placed on markers from chromosome 5 and 17, based on the report by Fukushima et al11 describing a translocation between 5q11.2 and 17q23 in a girl with Klippel-Feil anomaly and BDA1. Additional markers from Marshfield's sex averaged genetic map were examined. These included three markers from chromosome 17 (D17S1301, D17S1290, and D17S1303) and 14 markers from chromosome 5 (table 1). Each of the loci examined were individually amplified in 10 μl PCR containing 10 mmol/l Tris HCl (pH 8.3), 1.5 mmol/l MgCl2, 100-200 ng genomic DNA, 0.2 mmol/l dNTP, 0.12 μmol/l M13 tailed forward primer, 0.12 μmol/l reverse primer, 0.12 μmol/l IRD-700 labelled M13 forward(-29) primer (LI-COR, Lincoln, NE), and 1 Unit Taq enzyme. DNA was amplified over 16 cycles at 94°C for 30 seconds, 66-50°C for 30 seconds (−1°C per cycle), and 72°C for 30 seconds, followed by 23 cycles at 94°C for 45 seconds, 50°C for 30 seconds, and 72°C for 30 seconds. PCR products were size separated on 6% acrylamide gels, using the LI-COR DNA sequencer Model 4000 (LI-COR, Lincoln, NE) and analysed using RFLPscan software (version 3.0). Haplotypes were subsequently generated using Cyrillic software (version 2.1).
Lod score calculations for microsatellite markers on chromosome 5
Two point lod scores were calculated using MLINK, ILINK, and LODSCORE from the FASTLINK version12,13 of the LINKAGE software package.14 The LINKMAP program from the same software package was used to calculate location scores for multipoint linkage analyses. All lod and location scores were calculated using an autosomal dominant model, penetrance of 100%, and a disease frequency of 0.000001. Equal recombination frequencies between males and females were assumed.
RESULTS
Initial genotyping and linkage analysis of markers from chromosome 17 failed to yield evidence for linkage in the pedigree (data not shown). Lod scores greater than 3.0, however, were observed for two consecutive markers, D5S2848 and D5S1506, on chromosome 5p. Additional markers around this region were subsequently analysed, providing a maximum lod score of 6.91 at D5S477 (recombination fraction (𝛉) = 0.00) (table 1). Haplotype analysis provided evidence for an 11 cM critical region that cosegregates with the disease (fig 1). The distal end of this critical region was defined by an inferred recombination event in II.9 between markers D5S819 and D5S1986. The proximal boundary was further defined by an inferred recombination in II.5 between markers D5S1506 and D5S663. The location of the disease locus within the region between D5S819 and D5S1986 was further supported by multipoint linkage analysis (data not shown). As our initial linkage results were obtained before the report of Yang et al,4 the BDA1 locus at 2q35-36 was not evaluated for exclusion.
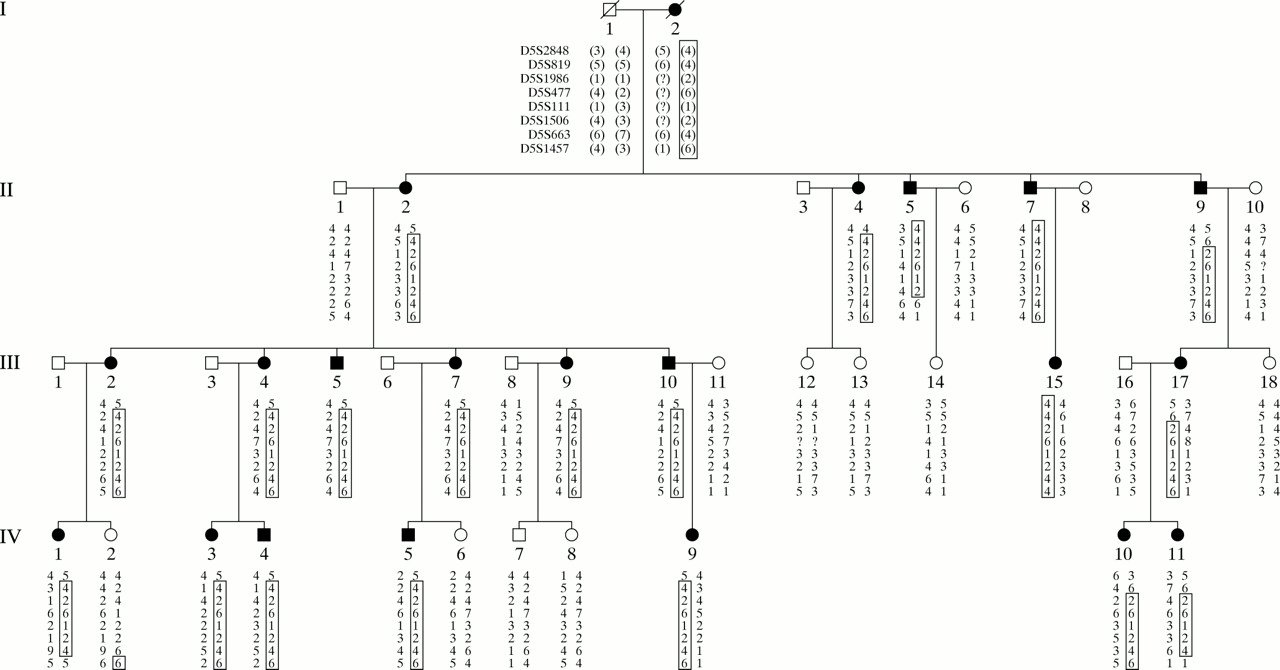
{kind=link}
Haplotype analysis of polymorphic markers from chromosome 5p in a family with mild brachydactyly. Filled symbols represent affected subjects. The disease haplotype is highlighted by the open boxes around the haplotypes.
There is a discrepancy between the position of our locus and the reported translocation breakpoint found in the girl with BDA1 and Klippel-Feil anomaly.11 If this girl's features are the result of a disruption in a gene at 5p13.2-13.3, the translocation may also be associated with a more complex, cryptic rearrangement. Similar phenotypic characteristics between the girl reported by Fukushima et al11 and our kindred include brachydactyly type A1 and wide spacing of the first and second toes. Although not reported, the radiographs in Fukushima et al11 also appear to show shortened fifth metacarpals, a feature also found in our kindred.
DISCUSSION
In this paper, we report a family with a relatively mild form of BDA1. Affected subjects were characterised by short middle phalanges of digits 2 to 5, a hallmark feature of BDA1, but lacked traits common in more severe forms, such as the absence of middle phalanges and terminal symphalangism.3 Other common findings in this family included shortened distal phalanges, a short fifth metacarpal, malformed epiphyses, and clinodactyly. Previous reports of families with brachydactyly have shown that subjects can inherit the disease as an isolated malformation15–18 or in association with other disorders. Slavotinek and Donnai19 described a boy with severe brachydactyly, valgus deformities, developmental delay, nystagmus, and scoliosis. Scoliosis was also described in a family with BDA1 and degenerative arthritis owing to discoid menisci,20 and in a family with brachydactyly, tall stature, and foot deformities.21 In the examination of affected members described in this report, there were no features observed other than BDA1.
We have identified a second BDA1 locus at 5p13.2-13.3 in a Canadian kindred, indicating that BDA1 is genetically heterogeneous. A previous report by Yang et al4 identified a BDA1 locus on chromosome 2q35-36 in two Chinese families. The families, while linked to the same region on chromosome 2, did not have a common haplotype within the defined interval. Our kindred had features similar to the families described by Yang et al4 although some differences in phenotype were noted. Many members in their study were completely lacking middle phalanges, a phenotype that was not observed in our family. Similarly, some subjects had shortened third metacarpals while this was not a marked feature in our affected patients. As originally described by Fitch,2 all hand bones may be proportionately reduced in size. For affected members in our kindred the distal and proximal phalanges as well as the metacarpals were often shortened below 2 SD compared to age related norms. The first proximal and fifth metacarpal were also frequently short.3 Yang et al4 described a number of features within their family that were apparently beyond Fitch's description, such as severely shortened distal phalanges, shortened third metacarpals, and shortened proximal phalanges of the fifth digit. These differences are not surprising given that different loci have been implicated.
Despite being the first disease described in terms of Mendelian autosomal dominant inheritance, a gene that causes BDA1 had not been identified until recently. During the preparation of this manuscript, Gao et al5 reported mutations of the Ihh gene in three Chinese families with BDA1. Having excluded PAX3 as the cause of the disease, Gao et al5 examined Ihh based on its position within the critical region on chromosome 2q35-36, and its involvement in regulating chondrocyte growth and differentiation during endochondrial ossification.22,23 Three different point mutations were identified, each causing amino acid substitutions in a portion of the protein predicted to interact with the Patched receptor.5 These mutations probably cause BDA1 through a haploinsufficiency of the wild type Ihh, resulting in decreased chondrocyte proliferation and attenuation of the negative feedback loop that regulates chondrocyte maturation during endochondrial ossification. Although Ihh mutations have been identified in people with BDA1, we are not aware of any genes within the chromosome 5 critical region that fit in the Ihh pathway. Despite this, there are a number of candidate genes in the 11 cM region in which we have found linkage. Two prime candidates include cadherin-6 and Npr3.
Cadherin-6 is part of a superfamily of proteins that mediate cell-cell interactions, some of which have been previously implicated in osteoclast differentiation.24–27 Experiments with dominant negative cadherin-6 isoforms suggest that cadherin-6 may be necessary for osteoclast differentiation by mediating heterotypic interactions between stromal cells and pre-osteoclast cells during osteoclast differentiation.28 This effect has not yet been shown in vivo.
Npr-3, a receptor involved in natriuretic peptide clearance, has been reported to be involved in mouse skeletal development. Homozygote Npr3 mutated mice have increased body lengths and longer digits because of aberrant proliferation in vertebral growth plates and delayed forepaw ossification.29 The delay was most marked in the hind and fore paws and was most noticeable at the extremities of the metacarpals and phalanges.
Our kindred show premature fusion of growth plates as well as malformed epiphyses resulting in clinodactyly. Fitch described BDA1 as “proportionate dwarfing of limb bones with the middle phalanges most affected as these are the last to ossify”.2 These patterns are very nearly the complete opposite to those seen by Jaubert et al.29 While Npr3 is implicated in an overgrowth syndrome, it is conceivable that a different mutation resulting in up regulation could give rise to the BDA1 phenotype seen in our kindred.
In summary, we have shown that BDA1 is linked to an 11 cM critical region on chromosome 5p13.2-13.3 in a four generation family affected with the disease. As previous studies have shown linkage of BDA1 to chromosome 2q35-36,4 linkage to a different locus in this study suggests that BDA1 is genetically heterogeneous. Further analysis of the 11 cM region is necessary to identify the gene involved.
Acknowledgments
The first two authors contributed equally to this work. The authors wish to thank L Racacho and H MacDonald for technical assistance. The authors also wish to thank the members of the family for participating in this study. DEB is a scholar of the Canadian Institute of Health Research.