Article Text

Statistics from Altmetric.com
Editor—Reports of patients with terminal de novo deletions of chromosome 15q26 are rare. Excluding cases of ring chromosome 15 formation with different sized deleted chromosomal segments, only seven cases with solely distal deletions of 15q have been published.1-7 All other cases resulted from unbalanced reciprocal translocations involving different chromosomes and are therefore not comparable with de novo terminal deletions as described in our case.
With two exceptions, all de novo cases had interstitial deletions between chromosomal bands 15q21-q25. Only the patients described by Roback et al 5 and Siebleret al 6 had terminal deletions of 15q26.1. The deletions in these patients were not investigated by FISH, but molecular genetic techniques showed the loss of one copy of the insulin-like growth factor 1 receptor gene. IGF1R is a tyrosine kinase containing transmembrane protein that plays an important role in cell growth control. It has been assumed that monozygosity for this gene, which maps to distal 15q26, will directly disturb this pathway and inhibit normal growth of patients.8
Today, in addition to classical cytogenetic banding methods, FISH techniques including comparative genomic hybridisation (CGH) can be used to provide a powerful tool to characterise chromosomal aberrations. In this study, we present the molecular cytogenetic findings and the detailed clinical phenotype of a girl with deletion 15q26.1 and compare these with other published cases. Our patient described here is, to the best of our knowledge, the second patient with a de novo terminal deletion at 15q26.1 and the first one well characterised by molecular cytogenetic techniques.
Case report
The female infant was the first child of healthy, unrelated parents. An ultrasound examination at 15 weeks of gestation showed intrauterine growth retardation. At 39 weeks of gestation a caesarean section became necessary because of fetal heart rate deceleration. The Apgar scores were 6, 8, and 10 at one, five, and 10 minutes, respectively. Her birth weight was 1980 g (<3rd centile) with a length of 42 cm (<3rd centile) and a head circumference of 30 cm (<3rd centile). The first chromosome analysis after birth in an outside laboratory showed a normal female karyotype. The girl had minor anomalies including micrognathia, low set ears, a broad nasal bridge, and a short neck (fig 1). Furthermore, there was a blood pressure difference between the upper and the lower extremities. Cardiac examination including cardiac catheterisation exhibited a complex heart defect with ventricular septal defect (VSD), atrial septal defect (ASD), preductal coarctation of the aorta, patent ductus arteriosus, and arteria lusoria. This complex congenital heart disease was corrected by several surgical interventions up to the age of 3 months. Laboratory findings including IGF1 and a screening for congenital infection were normal except for a transient hypothyroidism owing to maternal hypothyroidism. Renal ultrasonography showed a slight ectasia of the left renal pelvis from the age of 7 months. Neurological examination showed developmental delay but no other pathological findings. At the age of 15 months the infant could roll over but could not sit without support. Furthermore, she had severe feeding problems with gastro-oesophageal reflux and vomiting. Because of increasing vomiting and a lack of weight gain, a gastrostomy feeding tube had to be inserted. For the whole period of time the girl continued to have poor development and severe failure to thrive. At the age of 16 months her weight was 5300 g (<<3rd centile), length was 62 cm (<<3rd centile), and head circumference was 39 cm (<<3rd centile).
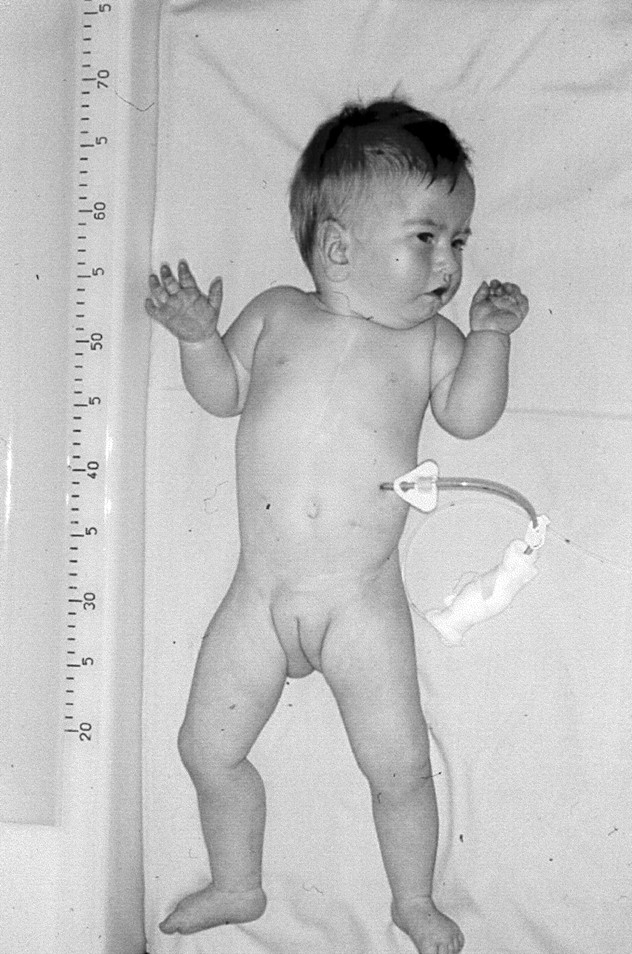
The patient at the age of 19 months.
Material and methods
Blood samples from the patient and her parents were drawn after informed consent. High resolution chromosome analyses from peripheral blood lymphocytes of the patient and both parents were performed using standard techniques. Preparations were GTG banded and karyotyped using the Ikaros system (Metasystems, Altlussheim, Germany).
Whole chromosome painting (WCP) was initiated using the probe for chromosome 15 (VYSIS). YAC clones for chromosome 15 were selected from the CEPH mega-YAC library and obtained through the Positional Cloning Centre at the Max-Planck Institute of Molecular Genetics (Berlin, Germany). YAC DNA was amplified and labelled by degenerate oligonucleotide primed polymerase chain reaction (DOP-PCR) with minor modifications.9 YAC-FISH was performed according to standard protocols. Hybridisation of commercial probes for the subtelomeric region of chromosome 15q (TelVysion 15q, VYSIS) and the all human telomeres probe (ONCOR) were according to the manufacturers' instructions. All probes used were directly labelled with fluorochromes.
Genomic DNA of the patient was investigated by comparative genomic hybridisation using normal male reference DNA as a control. DNA was isolated using standard methods. Briefly, genomic DNA samples were differently labelled by nick translation with SpectrumGreen®-dUTP (VYSIS, test DNA) and SpectrumOrange®-dUTP (VYSIS, reference DNA). For each hybridisation, 200 ng of labelled test DNA, 200 ng reference DNA, and 12.5 μg Cot-1 DNA were coprecipitated, resuspended in 14 μl hybridisation mix containing 50% formamide, 2 × SSC, and 10% dextran sulphate, denatured at 70°C for five minutes, and hybridised to denatured normal male metaphase spreads. Slides were incubated at 37°C in a moist chamber for two days. Post-hybridisation washes were performed as described previously.10 Images of the hybridised metaphases were evaluated using an epifluorescence microscope (Axiophot, ZEISS, Germany) fitted with different single band pass filter sets for DAPI, SpectrumGreen®, and SpectrumOrange® fluorescence. The microscope is equipped with a cooled CCD camera (Hamamatsu) for image acquisition. Image analysis and karyotyping (CGH) was performed using the ISIS analysis system (Metasystems, Germany). Diagnostic thresholds used for the identification of chromosomal under-representations (deletions) and over-representations (duplications) were 0.85 and 1.17.11
Microsatellite markers on chromosome 15q were analysed in the patient and her parents. Marker loci were chosen from the Généthon final linkage map and from the Marshfield comprehensive human genetic maps.12 13 Markers were amplified by PCR in a final reaction volume of 10 μl containing 10 mmol/l Tris, 1.5 mmol/l MgCl2, 100 μmol/l each dNTP, 0.4 U polymerase (Applied Biosystems), 7 pmol of each primer, and 20 ng of genomic DNA. One of the primers was end labelled with fluorescent dye. DNA amplification was carried out in an MJ Research PTC-225 thermal cycler. Reactions were electrophoresed on an ABI PRISM 377 automatic DNA sequencer (Applied Biosystems). Data were analysed using the computer programs Genescan v3.0 and Genotyper v2.5 (Applied Biosystems).
Results
Cytogenetic studies from the peripheral blood lymphocytes of the patient at the age of 9 months showed a female karyotype with a small deletion in the long arm of chromosome 15 at the 500-600 band level (fig 2).14 After conventional cytogenetics, the extent of the deletion was assumed to be from band 15q25∼26 to the distal end of the chromosome, but it was impossible to decide whether the deletion was interstitial or terminal. Maternal and paternal karyotypes were normal at the same resolution level.
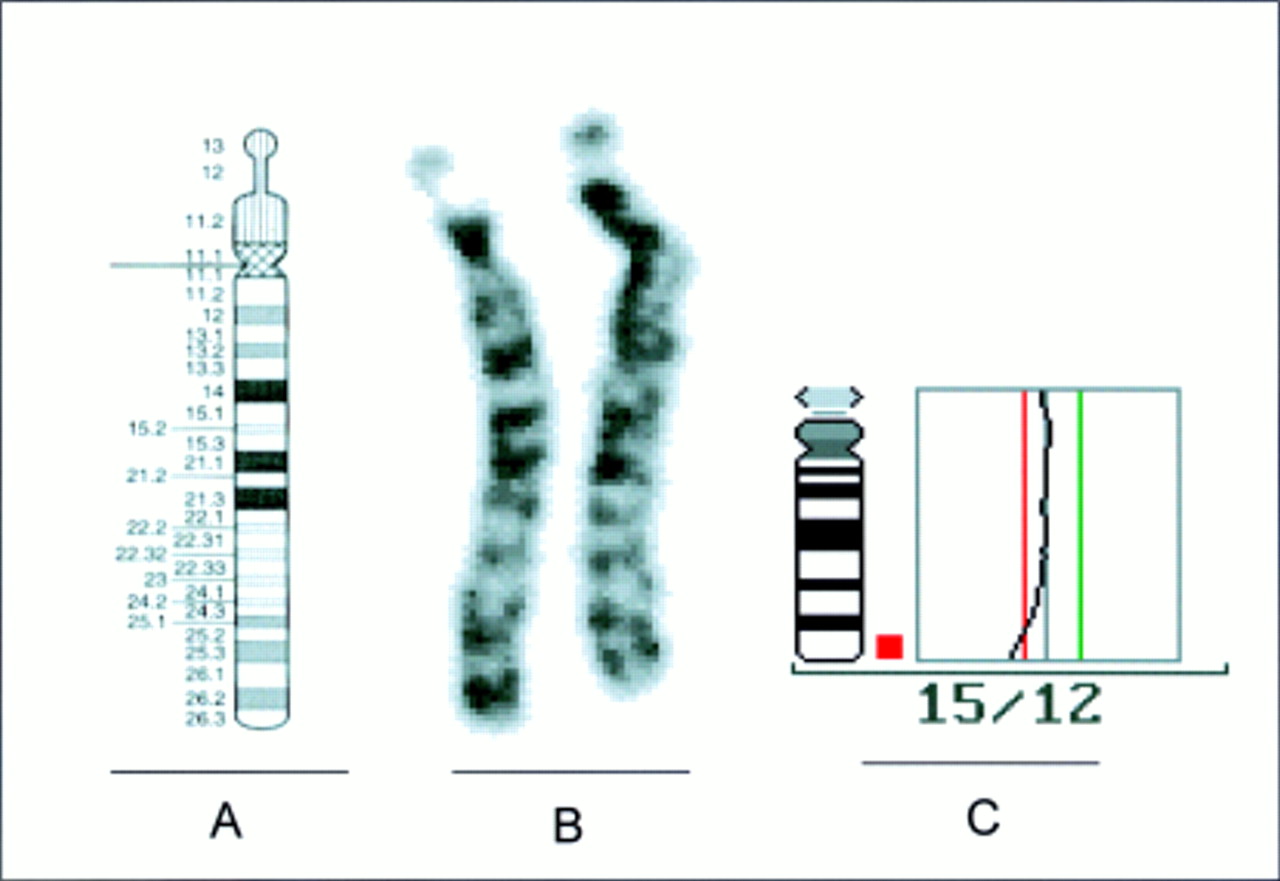
Ideogram14 of the human chromosome 15 (A) and the patient's chromosomes 15 (B) after GTG banding. The normal chromosome 15 is to the left of the deleted chromosome 15. (C) Averaged CGH ratio profile of 12 measured chromosomes 15 of the patient.
For further characterisation of the deletion, CGH was performed using total DNA from the patient as a probe. The averaged ratio profile analysis clearly indicated a terminal deletion (dim) of the chromosomal region 15q26 (fig 2). No other chromosome showed any ratio profile imbalance.
This result was in agreement with the FISH analysis using a chromosome 15 specific whole chromosome paint (VYSIS) showing homogeneous painting of the whole deleted chromosome 15 without any hint of a translocation of the missing chromosome 15 material to any other chromosome (data not shown).
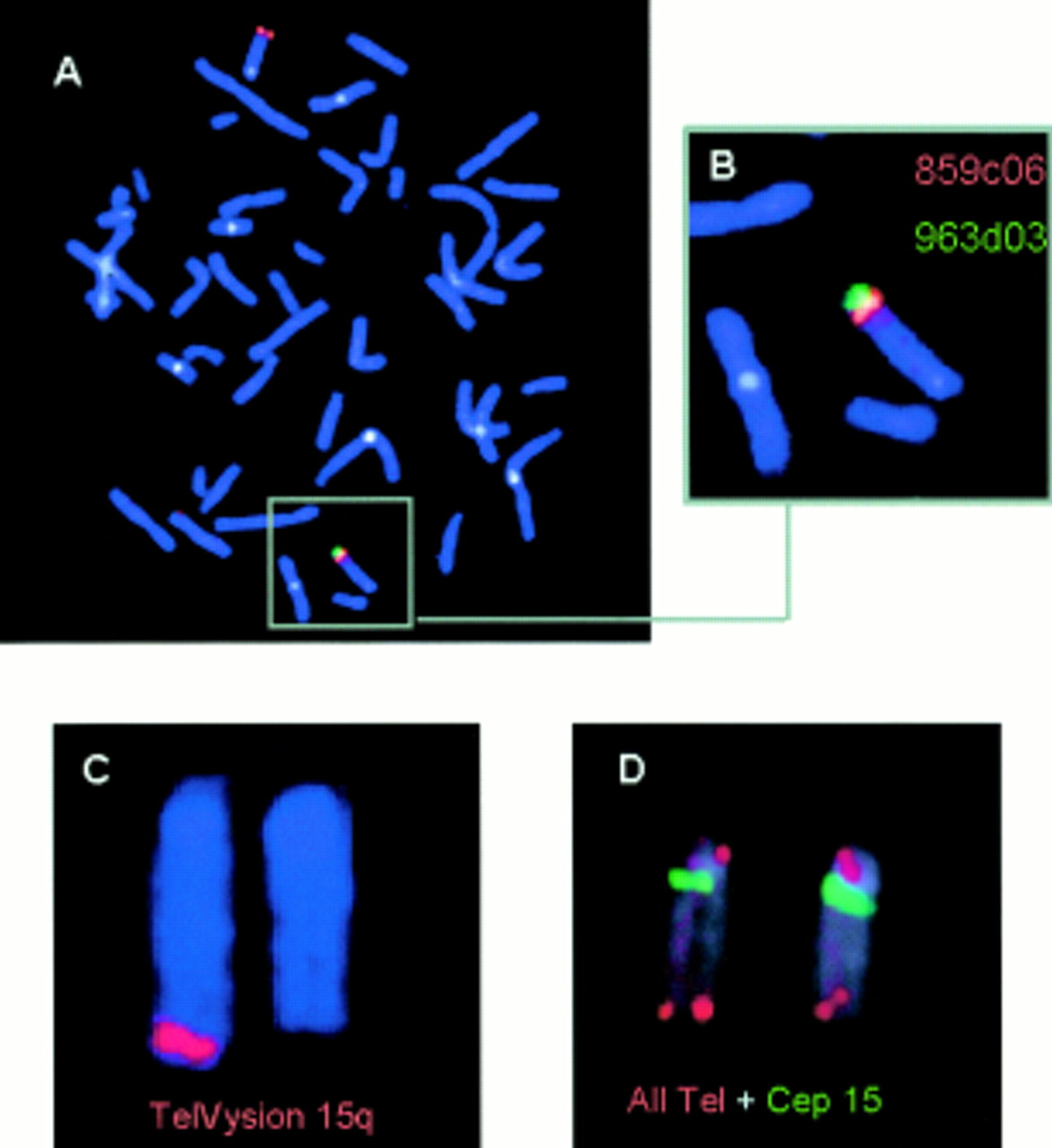
To define the proximal and distal boundaries of the deletion, FISH with different YAC clones was performed. Two of five YAC clones localised in chromosome band 15q25 (81-84 cM, table 1) showed signals on both chromosomes 15 on metaphase preparations of the patient (fig 3). Three YAC clones, 963d03, 895h10, and 882h08, localised distal to chromosome band 15q25 (98-110 cM), were missing from the patient's deleted chromosome 15 (fig 3).
Detection of chromosome 15q loci by FISH and microsatellite analysis
{kind=link}
{kind=link}
{kind=link}
FISH images of YAC clones and commercially available probes hybridised to the patient's chromosomes. (A) Fluorescence signals after hybridisation of the YAC clones 859c06 and 963d03. There is no signal for the latter clone in the patient's deleted chromosome 15. Both signals are seen in the linear orientation in the normal chromosome 15 (see B, magnification). (C) The subtelomeric TelVysion probe for chromosome 15q is also missing in the deleted chromosome 15. (D) A normal signal is seen for the all human telomeres probe detecting the highly repeated DNA (TTAGGG)n sequences located at the telomeres of all human chromosomes.
To delineate this chromosomal abnormality further, FISH with a probe hybridising to unique telomeric DNA sequences of chromosome 15q (TelVysion 15q, VYSIS) was performed. The investigation showed that a signal of this 100 kb sized probe for chromosome 15q is missing on the deleted chromosome 15 (fig 3). In contrast, FISH with an all telomeric probe (ONCOR) detecting the highly repeated (TTAGGG)n sequences located at the telomeres of all human chromosomes showed telomeric signals on both the normal and the deleted chromosome 15 as well as on all other chromosomes (fig 3). Thus the patient's karyotype can be summarised as: 46,XX, del(15)(q26.1).ish del(15)(D15S130−, D15S207/D15S157−, D15S120/D15S203−, D15S936−).
In order to complement the FISH data and to substantiate the loss of the IGF1R gene locus, a microsatellite analysis was performed. Twelve polymorphic markers from chromosome 15q were analysed (table 1). All the markers but those at D15S152, D15S1014, and D15S120 were informative for the family. Segregation of two different alleles clearly showed that the patient carries two copies of chromosome 15q proximal to D15S652 (table 1). Hence, the proximal boundary of the deletion is in the 10 cM interval between D15S652 and D15S130, so the deletion lies between D15S652 and the telomere. This finding is in accordance with the proximal boundary of the deletion defined by YAC hybridisation (table 1). Unfortunately, there is no true telomeric marker available on chromosome 15q, and the distance between the most distal marker at D15S642 and the telomere remains unclear. Additionally, it could be determined that the aberrant chromosome 15 was of paternal origin. TheIGF1R gene is located close to D15S120 as shown by radiation hybrid mapping between D15S107 and D15S87.15 16 These two markers are within the deleted region of our patient who therefore exhibits monozygosity for theIGF1R gene.
Discussion
Terminal deletions of chromosome 15q are rare events or are seldom diagnosed. Only a few cases of de novo distal deletions of chromosome 15q without ring formation have been described and the vast majority have been characterised by standard banding only yielding breakpoints in the range from 15q24 to 15q26. We describe here a new case of terminal deletion 15q26. Even with high resolution chromosome analysis, it was difficult to determine the exact size of the deletion. Therefore, we used different molecular cytogenetic approaches like CGH and FISH with YAC clones and commercially available telomeric probes to refine the deleted chromosome region to chromosome band 15q26. However, even with the molecular cytogenetic investigation, it was impossible to differentiate between an interstitial versus terminal deletion. The result of the FISH analysis with the YAC from the subtelomere of 15q (Telvision, D15S936) clearly showed a deletion on the aberrant 15 while a signal could be detected on both chromosomes 15 with the all telomeric repetitive probe (TTAGGG)n.
Therefore, it cannot be shown whether the telomeric sequence (TTAGGG)n at the distal end of the deleted chromosome 15 was from the paternal chromosome, or whether it derived from another chromosome by translocation. New studies on terminal deletions also suggest that de novo telomere addition could occur either mediated by telomerase or by recombination based mechanisms.17 In addition to the characterisation of the size of the deletion by in situ hybridisation, the deleted interval was determined by the analysis of microsatellites. These studies showed that the de novo deleted chromosome 15 was of paternal origin. This result is consistent with the paternal origin in the case described by Roback et al.5
Most patients with deletions of distal 15q have intrauterine growth retardation (IUGR), microcephaly, abnormal face and ears, micrognathia, a high arched palate, renal abnormalities, lung hypoplasia, failure to thrive, developmental delay, and mental retardation.5Apart from unbalanced chromosome translocations involving distal 15q and ring chromosome 15 syndromes, there are only seven previously described patients with de novo deletions of the distal long arm of chromosome 15.1-7 Most of these patients had interstitial deletions with different breakpoints indicating that the phenotypic discordance observed probably results from differences in the size and localisation of the deleted material.
Similarly to patients with distal deletion of 15q, many patients with ring chromosome 15 syndrome showed symptoms like IUGR, mental retardation, and microcephaly, but they more frequently had a triangular face, hypertelorism, café au lait spots, cryptorchidism, cardiac anomalies, and brachydactyly.18
To the best of our knowledge there are only two comparable cases to our patient with a deletion of 15q26.1 (table 2) that have been investigated by molecular genetic techniques.5 6 18These patients and our patient share intrauterine growth retardation, poor growth and development, and minor anomalies of the face. The female child described by Siebler et al 6 also had a triangular face and brachydactyly and exhibited characteristics of patients with ring chromosome 15 syndrome and deletion of 15q26.1. Renal malformations were only reported in the case of Roback et al 5 and our case. The patient of Robacket al also had lung hypoplasia, while our patient suffered from a complex heart defect. Feeding difficulties, as in our patient, were reported in four cases out of seven.
Only a couple of genes have been mapped to date in the distal part of chromosome 15, one of which is IGF1R (OMIM,http://www3.ncbi.nlm.nih.gov/htbin-post/Omim/getmap?chromosome=15q26). It has been proposed that haploinsufficiency of theIGF1R gene, which has been assigned to 15q25-q26,19 may play a role in the growth deficiency seen in patients with distal deletions of 15q25-26. Robacket al 5 refined the mapping ofIGF1R distal to 15q26.1 by deletion mapping. These findings were corroborated by Southern blot analysis of two patients with deletions of 15q26.1.6 TheIGF1R gene locus lies physically between the STS markers D15S107 and D15S87.16 Therefore,IGF1R is also deleted in our patient who displayed extreme pre- and postnatal growth retardation.
Peoples et al 16 investigated five children with de novo ring chromosomes 15 with breakpoints in 15q26.3 showing monozygosity of the IGF1Rgene in three of them. These three children had significantly more severe growth retardation in the first few years of life than one patient who retained the IGF1R gene on the ring chromosome. These data support a correlation between monozygosity for the IGF1R gene and severe growth retardation in early childhood, while patients who have retained two copies of the IGF1R gene show milder growth retardation.20
In vitro studies of fibroblasts of the two patients described by Siebler et al 6 showed that IGF1 receptor expression was decreased, while there was no evidence for impairment of the response to IGF1. Thus, Siebleret al 6 suggested that the growth retardation might not be related to monozygosity forIGF1R. However, the authors conceded that extrapolation from findings in skin fibroblasts to the situation in vivo is difficult.
De Lacerda et al 21 were the first to describe in vitro and in vivo studies of a patient with ring chromosome 15 syndrome and monozygosity forIGF1R. The female child showed prenatal and severe postnatal growth failure, a slightly triangular face, high arched palate, café au lait spots, and delayed psychomotor development. The patient's fibroblasts exhibited growth response in vitro to the addition of IGF1, similar to that of control fibroblasts. In contrast, the treatment of the child with short term recombinant human IGF1 (rhIGF1) caused no significant reduction in urinary urea nitrogen excretion, only 60% increase in calcium excretion, and no significant decrease in the GH secretion. Therefore, the authors suggested that the growth retardation could be the result of the absence of one IGF1R allele because of in vivo resistance to IGF1.
Studies on the effects of IGF1R in the cardiovascular system may support this assumption. These data showed evidence that IGF1 is an essential regulator of developmental growth and plays an important role in cardiovascular development.22 A variety of growth factors upregulate IGF1R on vascular smooth muscle cells and the data support the concept that IGF1R number per cell is an important factor for cellular growth response.
Therefore, monozygosity for IGF1R would be the best explanation for the complex heart defect seen in our patient. Thus, in addition to severe growth retardation, monozygosity forIGF1R might be a risk factor for the development of complex heart defects.
Acknowledgments
We thank the Max-Planck-Institute of Molecular Genetics, Berlin, for the YAC clones. The authors thanks Antje Gerlach and Britta Teubner for excellent technical assistance in the molecular cytogenetic experiments.