Article Text

Statistics from Altmetric.com
Editor—Around 1 in 1000 children is born or presents in early childhood with a severe hearing impairment.1 2 In developed countries, approximately 50% of these cases are attributed to genetic causes and the majority are non-syndromic with an autosomal recessive mode of transmission.3 Childhood onset non-syndromic sensorineural hearing loss (NSSNHL) is almost exclusively monogenic. This has facilitated the mapping of over 25 autosomal recessive NSSNHL loci (assigned DFNB) (Hearing Loss Homepage,http://hgins.uia.ac.be/dnalab/hhh). More recently, positional cloning and candidate gene screening strategies have led to the identification of 10 NSSNHL genes (Hearing Loss Homepage).
Material and methods
We ascertained a consanguineous family from the United Arab Emirates (UAE) comprising five subjects with severe-profound prelingual NSSNHL. Genomic DNA was extracted from peripheral blood samples using standard non-organic procedures. A genome wide search using the CHLC/Weber Human Screening set, version 8 (Research Genetics) was undertaken. PCR reactions were performed according to the manufacturer's instructions. This defined a ∼13 cM autozygous region on chromosome 2p23, delimited by the microsatellite markers D2S272 and D2S2347. The linkage interval incorporated the DFNB9 critical region (MIM 601071). Previously, mutations in the otoferlin (OTOF) gene were shown to cause NSSNHL at the DFNB9 locus (MIM 603681, table 1).4 5 More recently, however, further analysis of the OTOFtranscript has shown the existence of both short and long isoforms.6 Short isoforms are encoded by 28 exons.4 Long isoforms are derived from the alternative splicing of 19 additional 5′ exons.6 Analysis of the newly characterised 5′ exons in a DFNB9 family has led to the identification of the third OTOF mutation (table1).
Summary of known OTOF gene mutations in subjects with NSSNHL
Results
Following the original identification of shortOTOF isoforms as the cause of NSSNHL at the DFNB9 locus, we screened the 28 reported exons, 5′UTRsf (short form) and 3′UTR, in our family by direct sequencing. PCR products were purified on agarose gel and used as template with the BigDye Terminator Cycle Sequencing Reaction Kit (PE Applied Biosystems).7Two OTOF sequence variants were identified. The position of each variant is given with respect to the long isoform.6. Screening 5′UTRsf showed a small deletion (CCA) and insertion (G) variant located 137 bp upstream of the short isoform translation start signal (sf-137delCCAinsG). In addition, a G→C substitution was detected at nucleotide 2736 in exon 24. However, the 2736G→C variant does not change the leucine residue at position 912 (L912L). Both variants were homozygous in affected subjects, heterozygous in the parents, and not present in the unaffected family members. To investigate sf-137delCCAinsG further, we screened 18 UAE controls; the sequence variant was detected in 20 of 36 (56%) chromosomes. This included five controls homozygous for sf-137delCCAinsG. Based on this finding we concluded that sf-137delCCAinsG was a non-consequential polymorphic variant.
The failure to detect an OTOF mutation in our family and the observation that mutations in more than one gene have been identified at the same NSSNHL locus suggested the existence of a second NSSNHL gene at 2p23.8 9 However, following the report of a further 19 5′ exons encoding the long isoforms ofOTOF, we extended our mutation screen and identified a nonsense mutation in the otoferlin gene. A substitution of C→T in codon 237, spanning exons 8 and 9, changed an arginine residue to a stop codon (table 1, fig 1). The homozygous R237X mutation was detected only in affected family members and is predicted to encode a protein deficient in five of the six C2 domains.6 R237X was not present in 36 UAE control chromosomes. In addition, analysis of 10 white non-consanguineous sib pairs with NSSNHL, shown to segregate DFNB9 microsatellite markers, failed to detect the four knownOTOF gene mutations.

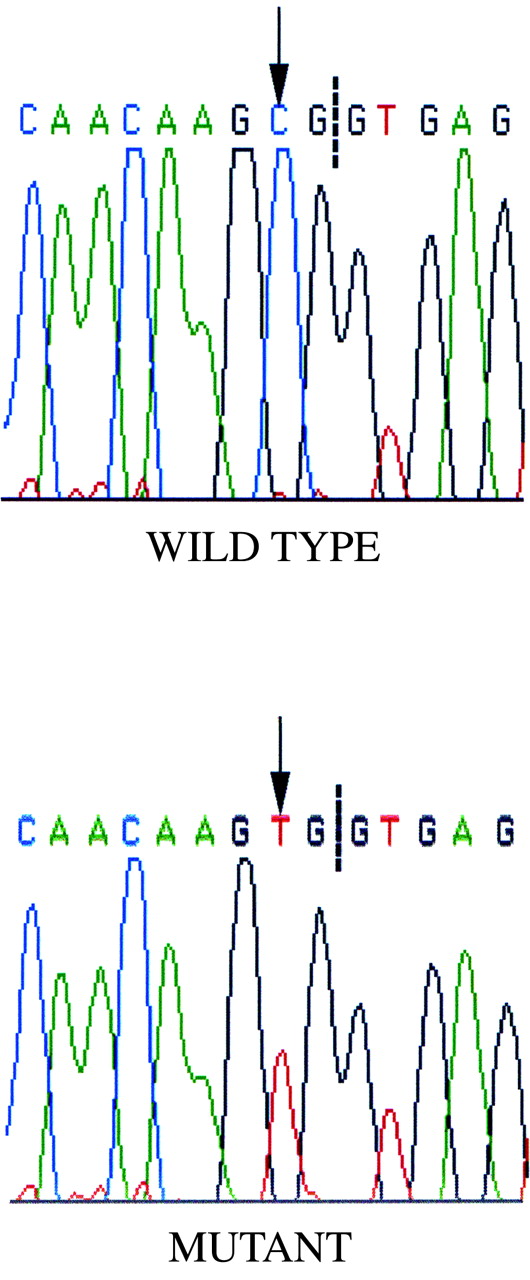{kind=link}
Sequence electropherograms illustrating the C→T base transversion (arrow) identified in the first nucleotide of codon 237. Splicing of exon 8 and exon 9 in the mutant mRNA produces a termination signal (CGA→UGA). The dashed line indicates the exon 8/intron 8 boundary.
Discussion
Our study has identified the fourth mutation in the otoferlin gene. More significantly, it represents the second mutation positioned within the newly characterised exons that encode the long isoforms. It will be of interest to define roles for the long and shortOTOF isoforms, given that two mutations affect both isoforms and two mutations affect just the long isoforms. Re-evaluation of the families involved may elucidate subtle clinical differences between the two subgroups. Our study also highlights the problems faced by researchers when no mutation is detected in a known disease gene within a family mapping to the same chromosomal location, especially in inherited disorders known to be highly genetically heterogeneous. Does the absence of a mutation necessarily suggest the presence of a second gene or is there a possibility that uncharacterised tissue specific transcripts exist? This reinforces the need for thorough assessment of new and existing genes in the tissues associated with the disease phenotype, as exemplified by the study of Yasunaga et al,6 and also the mechanisms by which they are regulated.
Acknowledgments
This work was supported by the NHS Research and Development Trust, Wellcome Trust, National Lotteries Charity Board through Defeating Deafness, and the European Union Framework V.