Article Text

Statistics from Altmetric.com
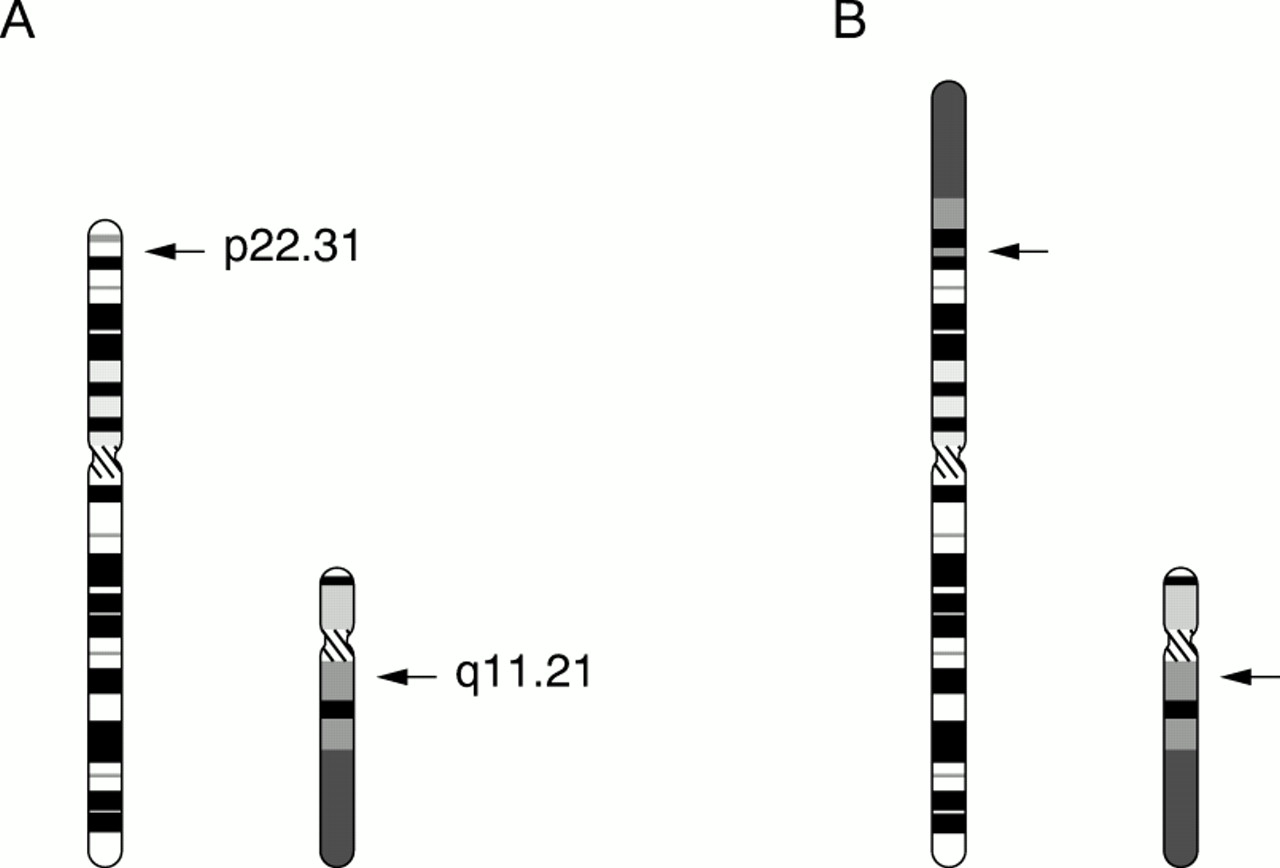
Editor—The genetic counselling of a pregnant woman who carries an Xp chromosomal deletion is far from straightforward. While the precise locations of the CDPX1(arylsulphatase E), steroid sulphatase (STS), and Kallman (KAL1) genes are known and FISH probes are available for these well characterised genes, the positions of putative mental retardation genes in this region have not yet been determined. Clinical and molecular studies undertaken over the past 10 years on patients with distal Xp deletions imply, however, that the putative X linked mental retardation (XLMR) gene,MRX49, lies distal toGS1 and STS but proximal to DXS31 and CDPX1 (fig1).1-4

(a) Case 6, (b) case 8, (c) case 9, (d) case 4, (e) case 12, and (f) case 13 of Ballabio et al,1 (g)-(j) cases BA16, BA20, BA139, and BA75 of Schaefer et al,3 (k) boy with IQ of 46, short stature, generalised ichthyosis, hypogonadotrophic hypogonadism, nystagmus, and photophobia,2 (l) boy with aggressive and hyperactive behaviour, myoclonic epilepsy, developmental delay, and no speech aged 4 years 8 months,4 (m) monozygous male twins with X linked ichthyosis, learning difficulties (LD), and epilepsy,10 (n) our patient, with short stature, Binder syndrome, and ichthyosis (consistent with the loss of the SHOX, CDPX1, and STS genes, respectively) but no significant learning difficulties. The presence (+) or absence (-) of LD is indicated for each case. A broken line indicates the chromosomal region within which the breakpoint is assumed to lie, while a solid line indicates a retained region.
Here we describe the clinical, cytogenetic, and molecular features of a boy with an unbalanced X;Y translocation resulting in a deletion of Xp extending from Xptel to the STS gene who, intriguingly, does not have learning difficulties (LD), despite the loss of this putative XLMR locus.
Case report
This 9 year old boy was delivered at term by caesarean section on account of fetal distress. He weighed only 2610 g but did not have any significant problems neonatally. His developmental milestones were achieved satisfactorily but he was investigated, aged 21 months, on account of his significant hyperactivity and ichthyosis. He has facial dysmorphism akin to that of Binder syndrome, including a broad nasal bridge and forehead, maxillary hypoplasia, relative prognathism, and dental malocclusion, in addition to terminal phalangeal shortening (fig 2). He suffered from epileptic seizures from the age of around 6 months, requiring prophylactic medication for two years. With the exception of one seizure that lasted 45 minutes, the fits were all brief and associated with pyrexia. His height lies just above the 3rd centile, while his head circumference and weight lie between the 25th and 50th centiles. He was diagnosed by a child psychiatrist as having attention deficit hyperactivity disorder (ADHD) and was treated successfully with methylphenidate.

(A) Radiograph of left hand showing shortening of distal phalanx of middle finger. (B) Dorsum of left hand. (C) AP and (D) lateral views of our patient, aged 9, showing facial dysmorphism similar to that in Binder syndrome (see text for description).
Psychometric testing, done by a senior educational psychologist, in addition to his assessment by his schoolteacher, indicated that he is of average cognitive potential and does not have any innate learning difficulties. His CT and EEG proved normal. Cytogenetic analysis, however, showed his karyotype to be 46,Y,der(X)t(X;Y)(p22.31;q11.21). The derivative X chromosome was found to lack the XYptel andSTS sequences but to contain a large region of Y long arm material, including the heterochromatic region and the XYqtel sequence (figs 3 and 4). Additional FISH and molecular analyses were carried out, localising the Xp breakpoint to between theSTS and KAL1genes (table 1). His mother is a carrier of the derivative X chromosome and has carrier levels of STS activity.

Chromosome analysis of our patient. (A) DAPI stained metaphase spread showing the presence of Y derived heterochromatin (arrow) on Xp. FISH analysis showing (B) the presence of the (red) XYqtel signal (arrow) at the tips of both arms of the derivative X chromosome and the (green) XYptel signal on the Y chromosome only, (C) the absence of STS signal on the derivative X chromosome, and (D) the presence of KAL1 signal (arrow) on the derivative X chromosome.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic indication of the origin of the derivative X chromosome showing (A) the X and Y chromosome breakpoints and (B) the derivative X chromosome.
Results of molecular (DXYS233, PABX, DXS996, DXS1118E, DXS6837, DXS1139, DXS6834, DXS1130, DXS237, DXS278, and DXS987) and FISH (XYqtel, XYptel, STS and KAL1) analyses undertaken to determine the location of the Xp breakpoint in our patient
All FISH probes were used essentially according to the manufacturer's instructions. Hybridisations were performed on metaphase chromosomes using STS or KAL1Xp22.3 region probe with DXZ1 chromosome X control probe (ONCOR)1 5 or chromoprobe T XYptel/qtel (CYTOCELL).6
Discussion
While the boy's short stature, craniofacial abnormalities, and ichthyosis are certainly consistent with the loss of theSHOX, CDPX1, andSTS genes, respectively, the severe hyperactivity which he exhibited in his early childhood was not readily predictable from his karyotype. Although both twin and adoption studies suggest that attention deficit and hyperactivity are strongly heritable,7 these complex disorders are likely to be multifactorial and genetically heterogeneous. The marked hyperactivity observed in this boy and the boy reported by Sprangeret al 4 might reflect the loss of an unidentified ADHD susceptibility gene in this Xp region, although we cannot exclude the possibility of the ADHD being an unrelated finding.
The results of the mapping studies (fig 1) have hitherto been interpreted as indicating that an XLMR gene is located betweenDXS31 and STS. These analyses have included a clinical and molecular study of 27 patients with deletions involving the distal short arm of the X chromosome,1 a description by Schaeferet al 3 of several patients with LD and terminal and interstitial Xp deletions,3 and a two point linkage analysis with X chromosomal markers on a family in which five males in two generations showed mild to moderate LD.8
The boy reported by Spranger et al,4 with an Xp terminal deletion with a breakpoint distal to the STS gene, was described as having LD in addition to short stature and chondrodysplasia punctata. Their molecular analysis would suggest that the putative MRX gene,MRX49, lies distal toGS1, which is consistent with the mapping data provided by Ballabio et al 1 (fig 1). Furthermore, very recently, a gene which resides between markers DXS1139 and DXS6837,VCX-A, was identified by further deletion mapping of 15 males with Xp deletions.9 This gene was reported to be deleted or retained in all of the subjects who had LD or were of normal intelligence, respectively.9
The lack of LD in the boy described here would suggest, however, either that the putative XLMR gene is located more proximally than previously considered or that if such a gene is located distal toSTS, its deletion is alone insufficient to cause LD. The genotype-phenotype correlation is, therefore, much less straightforward than might have been inferred from previous reports. This has important implications for the accurate counselling of carriers of similar Xp chromosomal deletions.