Article Text

Statistics from Altmetric.com
Editor—Since the first report by Mankinenet al 1 in 1976, over 30 cases with microscopically visible 1q deletions have been described. Patients with a distal deletion of 1q (q42 or q43→qter) have a recognisable pattern of malformations, albeit rather variable.2 3 The facial features include microcephaly, a full, round face with prominent forehead (sometimes with a metopic ridge), upward slanting palpebral fissures, epicanthic folds, a short, broad nose with a flat nasal bridge, thin lips with downturned corners of the mouth, micrognathia, apparently low set ears, and an abnormal palate (sometimes cleft). Patients are mentally and growth retarded, and may have variable cardiac, genital, and central nervous system anomalies. As most of these features suggest a chromosomal abnormality, patients with a deletion of distal 1q will usually be diagnosed after routine karyotyping. However, for submicroscopic distal 1q deletions, fluorescence in situ hybridisation with a 1qter specific probe will be required for the cytogenetic diagnosis. This test needs to be specially requested. In recent years, in situ hybridisation has led to the awareness that subtelomeric deletions below the level of the light microscope (<2-3 Mb) are a significant cause of malformation and mental retardation. One study identified previously undetectable abnormalities in 5% of 99 retarded patients.4 Whereas large deletions of distal 1q have been reported frequently, no submicroscopic distal 1q deletions have been described so far. Here we report the clinical and cytogenetic findings in two unrelated mentally retarded boys and one female fetus, one with a submicroscopic distal 1q deletion and the others with a partial submicroscopic trisomy of distal 13q in addition to a submicroscopic distal 1q deletion.
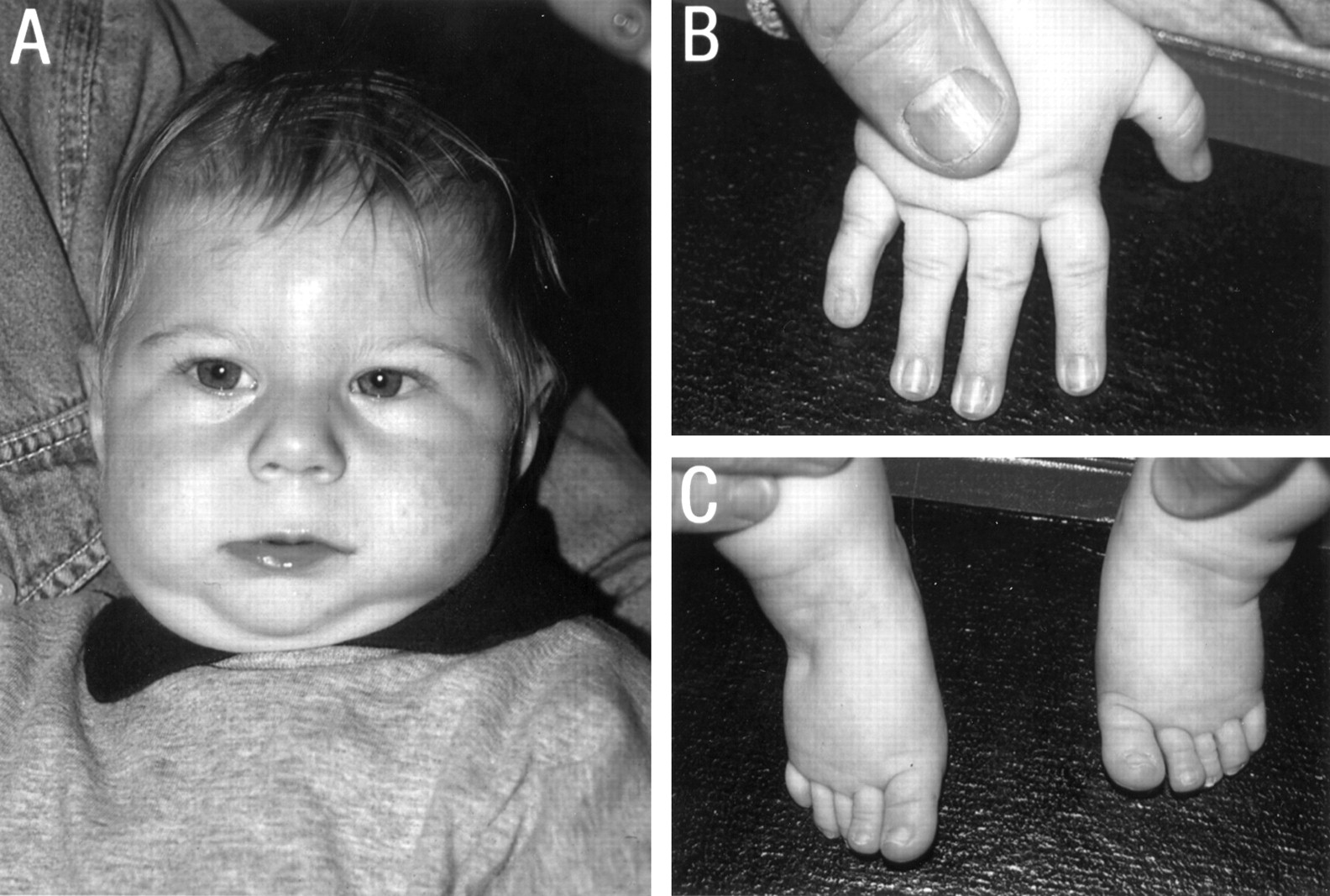
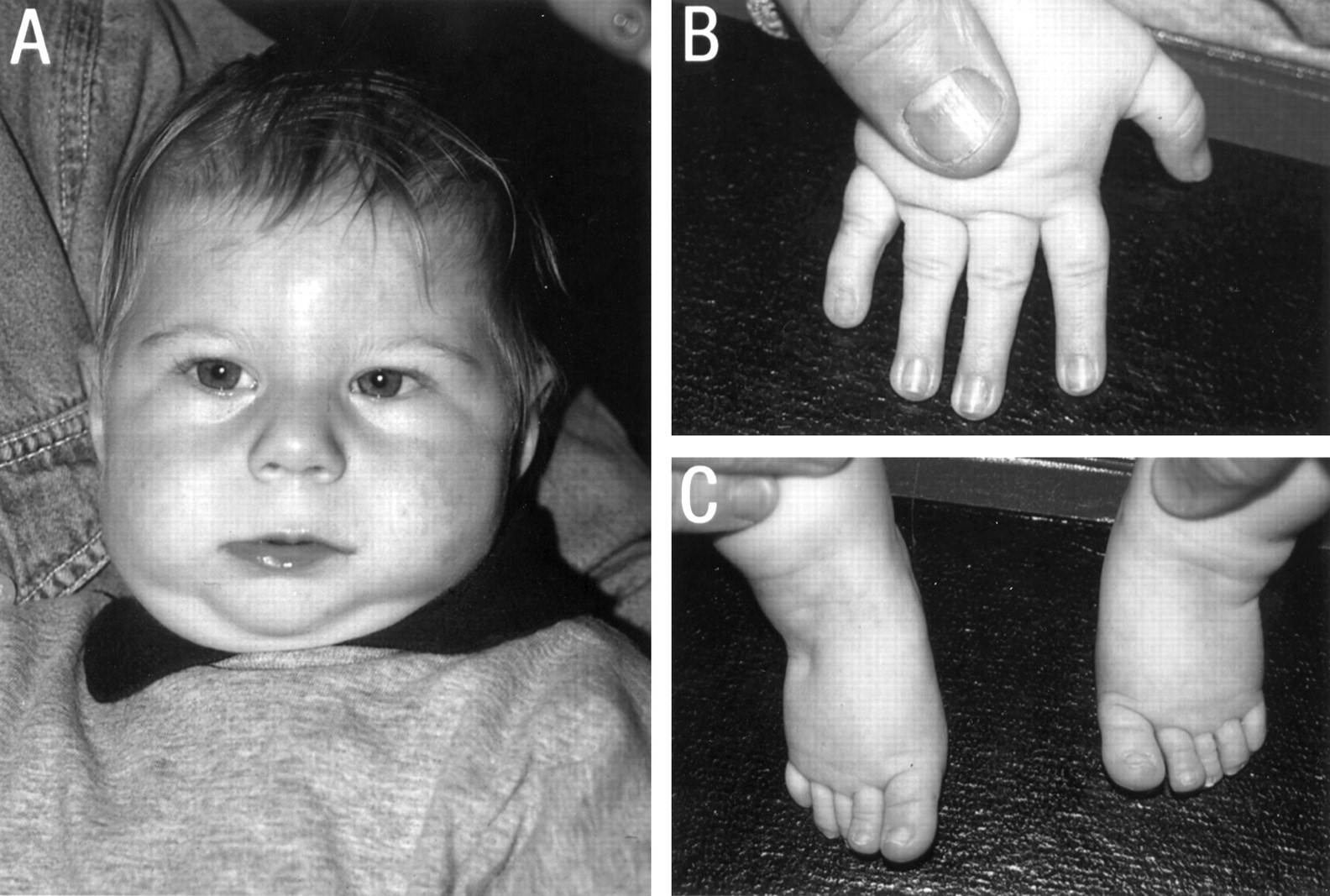
The first patient is a 7 year old boy who was born at 39 weeks' gestation with a low birth weight of 2300 g (<3rd centile). His 2 year older brother had a ureteric obstruction and his mother had had one miscarriage. There was bleeding in the eighth week of pregnancy for one week. At birth, he had hypospadias, a ventricular septal defect, and subaortic stenosis. His left kidney was non-functioning with vesicoureteric reflux of the right kidney. At the age of 2 months, he underwent a left inguinal herniotomy and a left orchidopexy. Early on, his visual behaviour was virtually absent and he began to use his eyes from 5 months of age. Several non-specific seizures were noticed in his first year for which he was treated with carbamazepine. No EEG abnormalities were seen. At the age of 17 months he underwent a fundoplication to control gastro-oesophageal reflux. The VSD was closed and a subaortic membrane resected at the age of 2½ years. An MRI scan of the brain showed partial agenesis of the corpus callosum with the posterior part of the body and the splenium missing at the age of 2 years 7 months. His development was severely delayed; he sat at 18 months, walked with support at 2 years, and did not develop any speech. At the age of 2½ years his head circumference was 41.5 cm (−5 SD) and his length 76.5 cm (<3rd centile). He had a full, round face, a short, broad nose with a broad base and a flat nasal bridge, and a long, smooth philtrum with a thin vermilion border (fig 1). Periorbital fullness, epicanthic folds, and strabismus were present. His ears had prominent, everted lobes. His fingers were tapering. G banded chromosome study showed a normal male karyotype, 46,XY. Testing for 7-dehydro-cholesterol was normal. Endocrine studies were normal.

Case 1 at the age of 2 years (A) and 6 years (B). Note the full, round face, periorbital fullness, epicanthic folds, strabismus, a short, broad nose with a broad base and a flat nasal bridge, long, smooth philtrum and thin upper lip, and prominent ears with everted lobes.
The second boy (case 2) was the first and only child of a non-consanguineous couple. The father had a normal daughter from a previous relationship and the mother was one of monozygotic twins. He was born at term with a birth weight of 2136 g (<3rd centile) and a head circumference on the 3rd centile. Ultrasound scanning at 20 weeks' gestation showed ventriculomegaly, micrognathia, and rocker bottom feet. Fetal blood sampling was performed in view of the above abnormalities and the karyotype was normal. By 28 weeks' gestation, the ventricular size was within normal limits. At birth, he had a cleft palate, small jaw, and hypospadias. Postnatal TORCH screen was negative. From birth, he was hypotonic with failure to thrive and frequent gastro-oesophageal reflux. A small patent foramen ovale was detected which did not require surgical treatment. On examination at the age of 16 months, he had developed severe microcephaly (head circumference 39 cm, −5 SD) with mild trigonocephaly and a metopic ridge. He had a full, round face, sparse, fine hair, periorbital fullness, and upward slanting palpebral fissures with epicanthic folds (fig 2). His nose was short with a long, smooth philtrum. He had a thin upper lip and downturned corners of the mouth with micrognathia and a cleft palate. His neck was short. On the right hand he had a single palmar crease and both hands were small with small nails. The feet were also small with overriding toes and rather oedematous dorsi of both feet. There was severe hypospadias and scoliosis. Testing for plasma amino acids and 7-dehydroxy cholesterol were normal. An EEG was normal. An MRI scan of the brain and spinal cord, at the age of 2 years, showed a rather poorly formed corpus callosum with a generalised paucity of white matter. The skeletal survey showed a hemivertebra at T11 and an anomalous vertebral body at T3. His development was severely retarded; at 22 months he was still not sitting and had no words. At the age of 2 years 5 months, his overall functioning was around the 8 month level.
Case 2 at the age of 1½ years. Note (A) full, round face, sparse, fine hair, periorbital fullness, upward slanting palpebral fissures with epicanthic folds, short nose with a long, smooth philtrum, thin upper lip and downturned corners of the mouth. and micrognathia. (B) Tapering fingers with small, narrow nails. (C) Small feet with overriding toes and oedematous dorsi.
The mother of case 2 had had a second pregnancy that was terminated at 17 weeks after structural abnormalities were identified in the fetus by ultrasound scan. Necropsy findings of the female fetus showed dysmorphic facies with a very small, receding mandible and a prominent maxilla and philtrum. There was a large midline cleft in the hard and soft palate, thick ear helices, and a flat occiput. The big toes were long and bulbous. A skeletal survey showed a hemivertebra at T7 in addition to the previous findings. A normal female karyotype was found in cultured amniotic fluid cells. The mother's twin sister had a severely retarded daughter with various dysmorphic features. She was macrocephalic (head circumference 57 cm at the age of 16 years, 98th centile) with a small mouth, mild microphthalmia, a high arched palate, small, overfolded ears, and a low posterior hairline. She had short fingers with clinodactyly and very small fifth toes. CT scan of the brain was normal.
FISH analysis using subtelomeric probes (under conditions described previously5-7) showed that case 1 had a de novo subtelomeric deletion involving the q arm of one of the chromosome 1 homologues (fig 3A). No other abnormality was detected. For case 2, subtelomeric FISH studies showed monosomy for the 1q subtelomeric region (fig 3B1) and trisomy for the 13q subtelomeric region. The additional copy of the 13q subtelomeric region is translocated onto the q arm of the derivative chromosome 1 (fig 3B2). This rearrangement was also present in the mother's second child, a female fetus aborted at 17 weeks. Subsequent FISH studies showed that both the mother and the mother's twin sister had balanced translocations of the 1q and 13q subtelomeric regions. The severely retarded daughter of the mother's twin sister had the contrasting unbalanced chromosomal rearrangement to her cousin (case 2); FISH showed that she is monosomic for the 13q subtelomeric region and trisomic for the 1q subtelomeric region.

{kind=link}
{kind=link}
{kind=link}
Subtelomeric FISH to metaphase chromosomes from (A) case 1 showing monosomy for 1q, (B1) case 2 showing monosomy for 1q, and (B2) case 2 showing trisomy for 13q. The p arm subtelomeric probe signals fluorescence red, whereas the q arm subtelomeric probe signals fluorescence green.
Microscopically visible telomeric deletions sometimes cause specific malformations and mental retardation syndromes such as 4p− (Wolf-Hirschhorn syndrome), 5p− (cri du chat syndrome), 9p−, 13q−, and 18p− syndrome and they can, therefore, be ascertained through specific phenotypes. Deletions of other subtelomere regions often have a less characteristic phenotype particularly for the submicroscopic subtelomere deletions. Recently, two other submicroscopic subtelomere deletions on autosomes have been associated with a distinctive clinical phenotype, 1p− and 22q−. Children with a distal 1p deletion are growth and mentally retarded with seizures, visual problems, large anterior fontanelle, asymmetrical and low set, dysplastic ears, deep set eyes, a depressed nasal bridge, a pointed chin, and fifth finger clinodactyly.8 9 22q telomere deletions are clinically associated with hypotonia, developmental delay, and absence of speech in the child.10 11
The two cases in this report support the presence of another recognisable submicroscopic telomere syndrome. Both patients shared a clinical pattern of severe mental retardation, growth retardation (prenatal onset), severe progressive microcephaly, hypospadias, corpus callosum abnormalities, cardiac anomalies, and gastro-oesophageal reflux. Facially, a short nose with a long, smooth philtrum, a thin upper lip, and full, round facies with periorbital fullness were present in both. These features are not restricted to submicroscopic 1q44 deletions as they have also been reported in patients with a microscopically visible distal deletion of 1q43→ter after routine G banding (table 1).2 3 Other clinical features individually present in the boys have also been reported in visible 1q43→ter deletions (table 1). Some of these features were also present in the 17 week old female fetus with the der(1)t(1;13)(q44;q34), such as micrognathia, cleft palate, prominent philtrum, and abnormal ears. Although the deletions in the present cases were submicroscopic, their phenotype fits into the spectrum of abnormalities observed in large distal deletions of 1q43→ter. This suggests that the “1q− phenotype” is mainly caused by hypoploidy of genes in the subtelomeric region, which is compatible with the fact that the subtelomeric chromosome regions are relatively gene rich.12 The clinical presentation, however, seems less severe in the submicroscopic cases, with regard to the motor development and life span, compared to the larger deletions. Obviously, this might be expected for a “contiguous gene syndrome”.
Clinical features in cases 1 and 2 compared to microscopic visible 1qter deletions
Several abnormalities in our three cases were related to the midline. This suggests that gene(s) involved in normal midline development might be located in the deleted region on 1q. The size of the telomeric deletion in two of the cases presented had previously been reported to be between 15.7 and 23.3 cM,7 which therefore defines the critical region for such a gene(s).
Although several clinical manifestations in the two cases can be observed in other chromosomal disorders, the combination of features seems to be distinctive: severe mental retardation, growth retardation (prenatal onset), severe progressive microcephaly, hypospadias, corpus callosum abnormalities, cardiac anomalies, gastro-oesophageal reflux, and a characteristic facies. Knowledge of the pattern of this “1qter− phenotype” will help clinicians to diagnose this chromosomal abnormality in their patients and to counsel the parents accordingly.
Acknowledgments
B B A de Vries was supported by a grant from the Ter Meulen Fonds (The Netherlands).