Article Text

Statistics from Altmetric.com
Editor—Familial adenomatous polyposis (FAP) is an autosomal dominant colon cancer predisposition syndrome in which patients develop hundreds to thousands of precancerous colonic polyps that have a high risk of becoming malignant.
Despite the fact that deletions of the APClocus were originally used to map1 2 and identify3 4 the APC gene, most studies of mutations in APC have used techniques which would not detect deletions.5-7 Molecular analysis of the APC gene in FAP patients has found the pathogenic mutation in 70% of cases,8 but a substantial minority have no mutation identified.
Interstitial deletions of chromosome 5q, which include theAPC locus, have been reported in patients with polyposis coli and mental retardation.9Submicroscopic deletions have been reported in only a few cases.10 11 In one family,10 linkage analysis with flanking and intragenic markers followed by in situ hybridisation with intragenic cosmid clones showed that the deletion was approximately 200 kb and included more than the 3′ half of theAPC gene and the 3′ adjacent D5S346 microsatellite. A recent report11 described a quantitative PCR assay to detect submicroscopic deletions which included the entireAPC gene and the adjacent D5S346 microsatellite in three unrelated Italian FAP families. These submicroscopic deletions do not appear to be associated with mental retardation.
Microsatellite analysis of families with FAP in our region showed one family with apparent non-maternity at D5S346 in one of the affected offspring of an affected mother, suggesting the possibility of anAPC deletion. A quantitative PCR assay was therefore developed to detect submicroscopic deletions of theAPC gene.
Previous analysis of 68 FAP families from the Yorkshire region found the pathogenic mutation in 46 cases (67%). These cases were referred for testing (with informed consent) from the Yorkshire Regional Clinical Genetics Service after clinical examination and counselling. All families with an unidentified mutation have been analysed by quantitative PCR; deletions have been identified in four families which is about 5% of the Yorkshire FAP families. In family 1, we studied three subjects affected with FAP. Cytogenetic analysis on one of the patients showed a normal female karyotype. In family 2, we studied four subjects affected with FAP. One of these patients, the father of the other three, did not develop the disease until he was 62 years old. In family 3, we studied two affected subjects. Only one sample was available from family 4. A patient with a cytogenetic deletion 46,XX,del(5)(q14.2q22.3) was also analysed by the quantitative PCR method.
Linkage analysis was performed using several flanking microsatellite markers: D5S299,12 D5S82,13DS5122,14 D5S346,15 MCC,16 and D5S318.17 Products were analysed by GeneScan analysis on an ABI 373A DNA sequencer with 672 software.
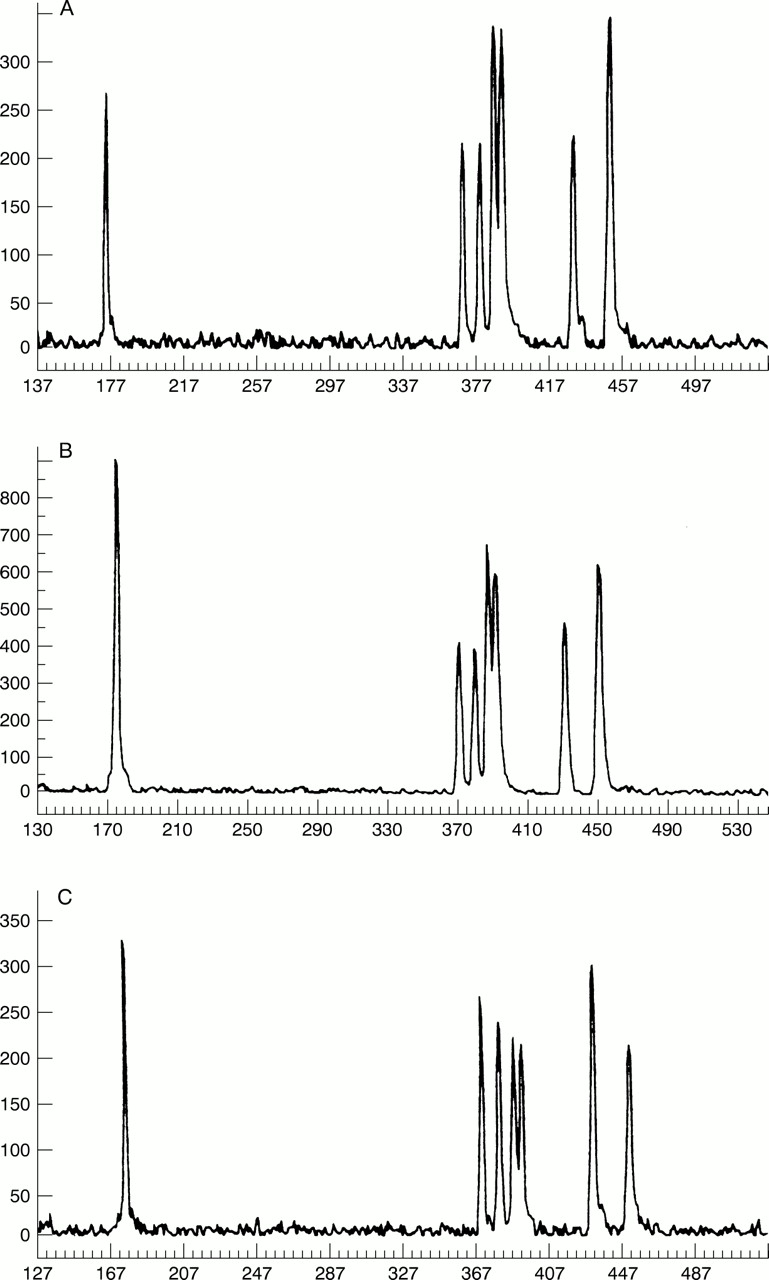
The quantitative multiplex PCR contained amplimers for exons 8, 15A, 15D, and 15F of APC 18 with exons 1, 2, and 3 of MPZ (myelin protein zero) (S C Yau, personal communication) used as a control (table 1). The 5′ end of one primer from each pair was labelled with the fluorescent phosphoramidite 6-FAM. The reaction was carried out in a total volume of 10 μl and contained 200-250 ng of template DNA, primers (table 1), 0.5 U of Promega Taqpolymerase, 200 μmol/l of each dNTP, 1.5 mmol/l MgCl2 in buffer (50 mmol/l tris, 15 mmol/l (NH4)2SO4, 50 mmol/l KCl, 0.085 mg/ml BSA). The reactions were performed in an MJ Research thermal cycler. Cycling parameters were as follows: 95°C for five minutes, 18 cycles of 95°C for 30 seconds, 52°C for 30 seconds, 70°C for one minute, and a final extension at 72°C for five minutes. Products were analysed by GeneScan analysis on an ABI 373A DNA sequencer with 672 software. Fig 1 shows results from two of our families and a subject with normal dosage for APC. The amplification of the test sample was compared with the amplification of a control sample with a known point mutation in a different part of theAPC gene. The peak height of each amplified exon was used to calculate dosage quotients, as described previously,19 for all amplicons. For example, the dosage quotient (DQ) for exon 8 of APC and exon 1 of MPZ is (sampleAPC exon 8 peak height/sampleMPZ exon 1 peak height)/(controlAPC exon 8 peak height/controlMPZ exon 1 peak height). EachAPC exon was compared with eachMPZ exon. A dosage quotient close to 1.0 indicated that two copies of the test gene are present; a dosage quotient close to 0.5 indicated only one copy. Dosage quotients and standard deviations of the mean dosage quotients were calculated using duplicate test and control sample loadings.
Primer sequences
{kind=link}
Electrophoretograms of quantitative PCR of exons 8 and 15 of APC and exons 1, 2, and 3 of MPZ. From left to right the peaks are exon 8 APC, exon 15A APC, exon 15D APC, exon 1 MPZ, exon 2 MPZ, exon 15F APC, and exon 3 MPZ. (A) Family 2 deleted for exons 8, 15A, 15D, and 15F of APC and D5S346. (B) Family 4 deleted for exons 15A, 15D, and 15F. (C) Normal (point mutation in APC identified).
Dosage quotient and standard deviation results are shown in table 2. The values were close to expected values with standard deviations approximately 10% of the mean dosage quotient. Two clear groups of results emerged with no overlap seen between values close to unity or values close to 0.5. Results indicating deletions (dosage quotients close to 0.5) were seen in each case with more than one amplicon, excluding allelic dropout as a cause of reduced amplification.
Dosage quotients (DQ) and standard deviation (SD) calculations for electrophoretograms shown in fig1
Three of the four deletions identified were distinct. Family 1 showed apparent non-maternity with marker D5S346 between the affected mother and one of her affected daughters. Quantitative PCR showed the affected subjects to have only one copy of exon 8, exon 15A, exon 15D, and exon 15F of the APC gene and we concluded that the mutation in this family was a deletion which extended from at least exon 8 of APC to the 3′ D5S346 microsatellite. A similar deletion was identified in family 2. In this family, all affected subjects were homozygous for the same MCC allele which suggests that the deletion could include this locus. In family 3, the deletion extended from at least exon 8 to exon 15F ofAPC. Microsatellite analysis showed the proband to be heterozygous at D5S346 and D5S82. In family 4 dosage quotients for exon 8 of APC showed that there were two copies of this exon present but only one copy of the regions of exon 15 examined. The proband was heterozygous at D5S346. We concluded that the mutation in this family was a deletion which included exons 15A, 15D, and 15F, but did not extend as far as D5S346. A summary of the microsatellite data and quantitative PCR is shown in table 3.
Summary of microsatellite and quantitative PCR results
Using a quantitative PCR assay we found submicroscopic gene deletions of APC to be the pathogenic mutation in four unrelated FAP families in Yorkshire, accounting for about 5% of our FAP families. Taken together with the report of De Rosaet al 11 who found submicroscopic deletions in three Italian families with FAP (17% of their FAP pedigrees), these data suggest that submicroscopic deletions of the APC gene may be more common than is evident from published reports, perhaps because they would be undetected by conventional mutation scanning methods. For example, a recent study7 found APCmutations in 106/190 FAP families but did not search for gene deletions. Our evidence suggests that gene deletions may be found in a substantial minority of FAP patients. That at least three of the four submicroscopic deletions were distinct, differing in the loss of D5S346 and exon 8, eliminates an undue bias in our sample because of a common founder mutation. As the assay is relatively quick and easy to perform, it will now form part of our initial diagnostic mutation screen for a new FAP patient.
A number of quantitative methods have been described to measure gene dosage, including Southern blotting,20-22 oligonucleotide hybridisation, competitive PCR,23-25 and more recently Taqman assays26 and comparative genomic hybridisation (CGH) either using metaphase spreads27 or arrayed targets.28 Any useful dosage assay should meet criteria of specificity (absence of false positives), sensitivity (absence of false negatives), reproducibility, and be reasonably economical in time and material. Hybridisation assays using genomic DNA digests are somewhat more time consuming than PCR based methods and usually involve the use of radiolabelled probes. CGH is a promising alternative, but limitations in the size of deletions or duplications detectable by metaphase spreads must be overcome by the availability of arrayed DNA before the technique can be generally applicable. Since automated fragment analysis is already widely used in genetic diagnostic laboratories, the use of dosage quotient analysis offers a robust and accessible means to test for microdeletions and duplications.
Pathogenic duplications or deletions have long been reported, from α globin23 and dystrophin29 to tumour suppressor genes includingBRCA1,30 BRCA2,31 andhMSH2.32 It is therefore important that genetic testing includes the detection of altered gene dosage where appropriate as conventional mutation scanning methods may miss submicroscopic deletions.
Acknowledgments
We thank Michael Yau, NE Thames Regional DNA Laboratory, Guy's Hospital, London for providing primer sequences for STSs from theMPZ gene.