Article Text

Statistics from Altmetric.com
Editor—Uniparental disomy (UPD) is the inheritance of both copies of a chromosome or a chromosomal region from one parent.1 2 When both chromosomes from one parent are present the phenomenon is called “heterodisomy”, whereas the presence of two copies of only one of the parental chromosomes is called “isodisomy”. A map of UPD for the human genome has been proposed and covers several areas of the 22 autosomes and the X chromosome.1 3 Not unexpectedly, growth abnormalities and other endocrine disturbances appear to be frequent in patients with UPD for whole chromosomes or even for small chromosomal regions.3 Although UPD appears to be rare among patients with various congenital defects,4 disomic human sperm nuclei are present relatively frequently and without interchromosomal differences.5 The true incidence of UPD may be significantly higher if the various molecular and cytogenetic possibilities for disomy are considered, and the genome is tested by densely located polymorphic markers.1-5
UPD for chromosome 2 has been reported in five patients,2three cases of heterodisomy and two of isodisomy.6-10 No particular clinical phenotype was seen in one of the patients,7 but intrauterine growth retardation (IUGR), oligohydramnios, and pulmonary and genital hypoplasia were present in all of the other cases.6 8-10 Additional manifestations in the individual patients were probably the result of the cytogenetic event that preceded UPD; two of the patients had confined placental mosaicism for trisomy 29 10 and another had trisomy 2 mosaicism in amniotic fluid culture.6
In the context of genetic studies for the mapping of Carney complex (CNC), a normal subject was found to have only maternal alleles of two polymorphic markers mapping to the short arm of chromosome 2. The absence of a microdeletion or other chromosomal anomalies (by classic and molecular cytogenetics) suggested that the patient had maternal isodisomy for a segment of chromosome 2. This region is at least 100 000 base pairs (bp) long,11-13 lies between polymorphic markers D2S123 and CA2, and corresponds to cytogenetic band 2p16.11 12 It is known to be frequently involved in chromosomal and molecular aberrations in endocrine and other types of tumours.11-13
Our patient was a normal 22 year old female; one of her sisters and possibly her father are affected with Carney complex (CNC), a familial multiple endocrine neoplasia and lentigenosis syndrome. The patient herself was a healthy woman (fig 1), who participated in a genetic study regarding CNC to discover if she is a carrier for that syndrome. For this, and all the studies described here, the patient signed an informed consent form under study 95CH059 approved by the Institutional Review Board of the National Institute of Child Health and Human Development. Her medical history was unremarkable. The patient was born weighing 3374 g after a term pregnancy and normal delivery. She had recurrent urinary tract infections as a child and she was on preventive antibiotics for this problem. She had menarche at the age of 11 years; adrenarche and thelarche were also reportedly normal. Her final height was 1.57 m, which was not different from that of her family's; obesity, acne, and hirsutism were problems that developed after puberty. Because of her family history, the patient was screened for Cushing syndrome but the investigation was negative (data not shown). A detailed physical examination showed no abnormalities, with the exception of mild obesity.

(A, B) Pictures of the patient; there are no dysmorphic features or any other abnormalities. (C, D, E) D8S284, D5S421, and D9S175 polymorphic marker analysis, respectively, for confirmation of paternity: The order of the lanes (from the left) is: father, mother, and patient. There is appropriate biparental inheritance of the two alleles.
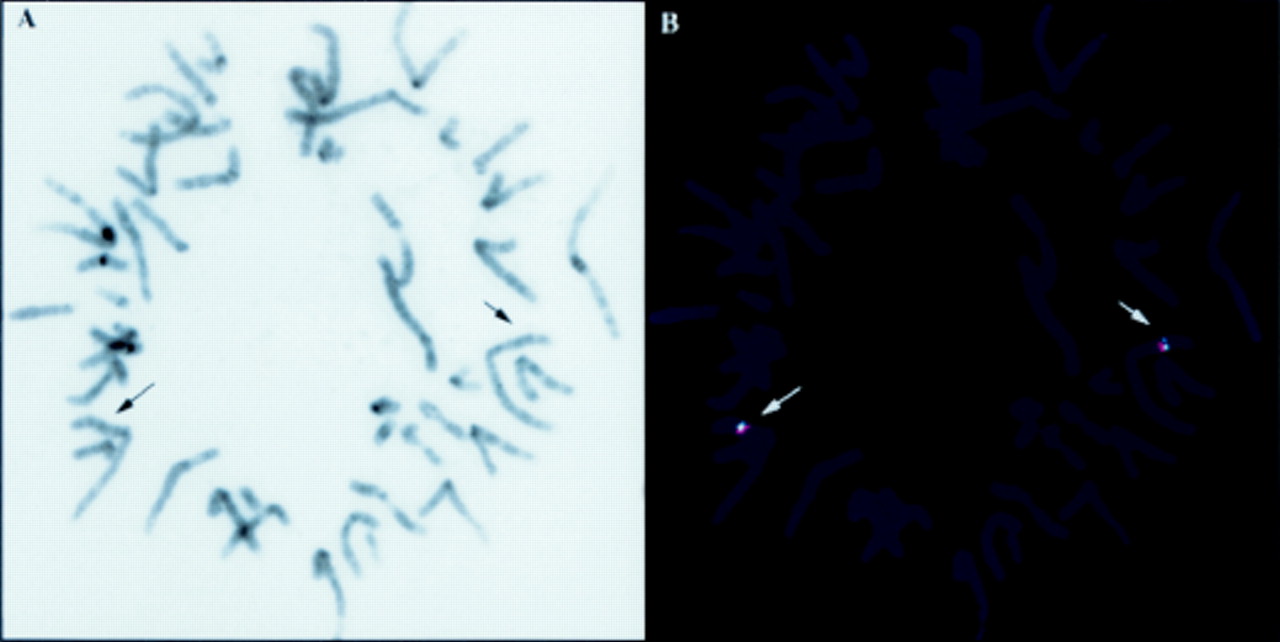
DNA was extracted from peripheral blood as previously described.11 Polymorphic markers around the CNC locus on chromosome 211 12 were tested by the polymerase chain reaction (PCR), following end labelling with γ-32P ATP of the reverse primer for each marker (figs 1 and 2). Markers on other chromosomes were tested for confirmation of paternity; three are shown in fig 1: D8S284, D5S421, and D9S175. The sequences and genomic order of these primers are available in the genome database on line (http://gdbwww.gdb.org/ and http://www.genome.wi.mit.edu). High resolution karyotype analysis was obtained by standard methods; a total of 25 cells were examined. Fluorescence in situ hybridisation (FISH) was performed with two bacterial artificial chromosomes (BACs) as probes, as previously described14 15 (fig 3). The BAC addresses were 79-H-12 (which contains polymorphic marker D2S391) and 43-E-19 (which contains polymorphic markers D2S123 and CA2). Both clones were obtained from a commercially available library (Research Genetics, Huntsville, AL). Location, primers, and PCR conditions for these two markers are available on line (see above) or have been published before.11-13 16 Details on these two BACs and their location in the physical map of chromosome 2 have been published elsewhere.12 16 Using nick translation, BAC 79-H-12 was labelled with biotin-16-dUTP (Boehringer Mannheim Co, Indianapolis, IN); BAC 43-E-19 was labelled with digoxigenin-11-dUTP (also from Boehringer Mannheim Co, Indianapolis, IN). Both probes were simultaneously hybridised on metaphase chromosomes that were prepared from the patient's peripheral lymphocytes, as previously described.17 18
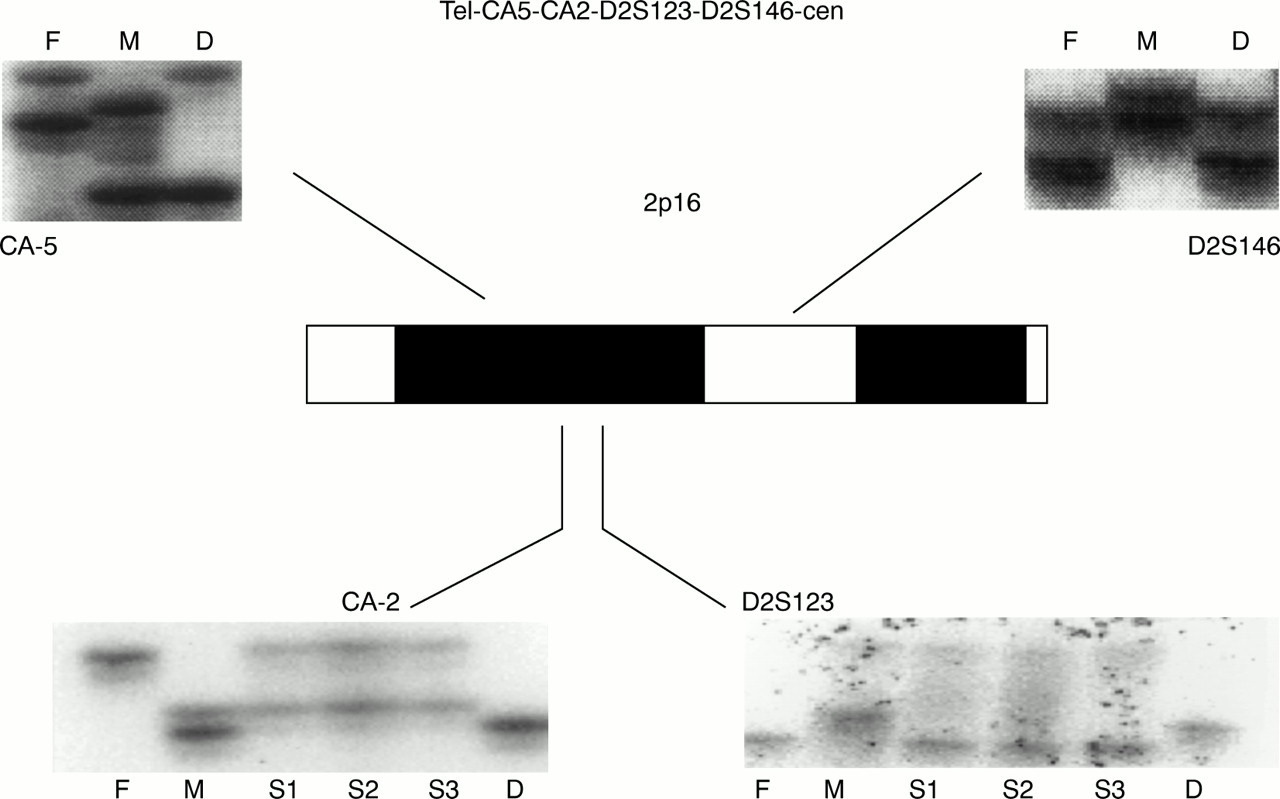
Ideogram of the 2p16 region and genotyping for four polymorphic markers mapping to that region in the order shown (tel-CA5-CA2-D2S123-D2S146-cen). Whereas for CA5 and D2S146 there is appropriate biparental origin of the two alleles, for markers CA2 and D2S123 the patient lacks the paternal alleles; instead, she is a homozygote for one maternal allele, suggesting maternal isodisomy for this chromosomal segment. F=father, M=mother, D=daughter (the proband), S1, S2, and S3=the three sibs of the patient.
{kind=link}
{kind=link}
{kind=link}
Two specific hybridisation signals were detected on both chromatids of chromosome 2 at band 2p16; no other hybridisation sites could be detected. (A) Inverted DAPI image for a representative metaphase spread (giving a G band-like appearance); arrows point to 2p16. (B) FITC and TRITC images of that same metaphase spread superimposed on the DAPI image; the arrows point to the hybridisation signals of the two probes, one of them for the D2S123/CA2 locus and the other for the D2S391 locus (near CA512 16). There are no deletions or other rearrangements involving the locus on the patient.
Our results indicated that for two markers on chromosome 2, CA2 and D2S123, the patient had only maternal alleles (fig 2). For both markers, the mother of the patient was heterozygous, whereas the patient was homozygous for one of the maternal alleles. The absence of a clinical diagnosis of CNC was consistent with the absence of a paternal contribution for this segment of the genome where CNC had been mapped. Fig 2 also shows an equal maternal and paternal contribution for the first informative markers on either side of the CA2/D2S123 locus,12 16 CA5 on the telomeric side (approximately 1 cM from CA-2/D2S123) and D2S146 on the centromeric side (approximately 4 cM from the CA2/D2S123 locus). Paternity was confirmed by the testing of other polymorphic markers localised on other segments of chromosome 2 or on different chromosomes (figs 1 and 2). For all informative markers tested, there was equal paternal and maternal contribution, as expected. A high resolution karyotype was obtained from the patient's peripheral lymphocytes; it did not show any abnormalities on chromosome 2 or on any other chromosomes. FISH with BACs previously identified as containing 2p16 genomic material,12 16 including one clone (43-E-19) that contained the markers D2S123 and CA2, showed the expected two signals from each of the chromosomes 2 of the patient (fig3). There were no structural abnormalities of 2p16 detected by these two probes and the intensity of the signal from all positive alleles was identical for all spots.
UPD for chromosome 2 has been reported in five patients.1 2 6-10 In all cases, only maternal UPD was observed and it was present across the entire length of the chromosome.1 2 As in the reported cases, our patient too had maternal UPD; however, this was only confirmed for a short segment of chromosome 2, around markers D2S123 and CA2. Other chromosomal defects (microdeletion and rearrangements) were excluded by both classical and molecular cytogenetics. The region harbouring D2S123 and CA2 is at least 100 000 bp long.12 13 However, the exact length of UPD in the reported patient is unknown, since there are no other known informative polymorphic markers in a region that extends for 4-5 cM around D2S123 and CA2.12 16 The patient did not have UPD for markers CA5 and D2S146 (fig 2), which are located several million bp telomeric and centromeric, respectively, of the CA2/D2S123 locus.11-13 16
UPD, whether it is iso- or heterodisomy, is usually observed along the entire length of the chromosomal arms involved.1-3 One exception is that of chromosome 11 in patients with Beckwith-Wiedemann syndrome (BWS); they often have germline or somatic UPD for a segment of the short arm of chromosome 11 around the BWS locus on 11p15.19 20 However, as more patients are tested for polymorphisms of known genes, anonymous sequences, or polymorphic markers, UPD is discovered for smaller areas of the genome, surrounded by regions for which appropriate, biparental inheritance can be shown. One recent example is that of a patient with autosomal recessive abetalipoproteinaemia caused by a mutation in the microsomal triglyceride transfer protein gene; he had UPD for a 150 000 bp long segment of chromosome 4q.21
Our patient did not have any detectable clinical abnormalities. In general, UPD may cause clinical symptomatology, if the disomic area harbours imprinted genes or genes that harbour mutations that do not cause disease in the presence of a normal functioning allele.1-3 Whether UPD for chromosome 2 has any clinical consequences remains debatable; of the five patients reported, one was normal,7 whereas the others had significant findings, including IUGR, oligohydramnios, and pulmonary and genital hypoplasia.6 8-10 The cytogenetic events that preceded UPD, such as mosaicism for trisomy 2,6 9 10 may be responsible for some of these clinical manifestations. Nevertheless, the finding that maternal UPD for 2p16 did not cause any clinical problems to our patient, defines a region on chromosome 2 that appears to be free of imprinting effects.
The CA2/D2S123 locus has been shown to be involved in loss of heterozygosity (LOH) in several neoplasms, including thyroid and adrenal tumours.22 23 However, the facts that UPD of this region may be associated with normal clinical history and that, in general, segmental UPD may be present at the somatic level only20 24 25 makes the interpretation of LOH studies of the CA2/D2S123 locus difficult. Such LOH may be real, only if heterozygosity is shown in the non-tumourous parts of the same tissue.
It is noteworthy that the CA2/D2S123 locus is also frequently involved in microsatellite instability in human tumours.11 26 Our data indicate that this, and one proximal to the CA2/D2S123 locus region, have a high recombination rate and perhaps harbour a fragile site.11 12 16 UPD occurs as a result of a meiosis type I, meiosis type II, or a mitotic error (for proven post fertilisation defects).1-3 20 Thus, UPD may occur more frequently for regions harbouring unstable or highly recombinant regions or areas of chromosomal attachment during pairing. The mechanism that led to our patient's defect is unknown and may be any of those suggested for UPD,1-3 20 24 25 including partial trisomy rescue.6 9 10 Trisomy 2 may be incompatible with life27-29 and, similarly, most duplications of segments of chromosome 230-35 (leading to partial trisomy for that chromosomal region) or deletions cause serious problems.36-38
In summary, we present a normal subject with segmental UPD for 2p16. This case has implications for the mapping of potentially imprinted genes on chromosome 2 and tumour studies involving 2p16. It also shows that small regions of UPD in humans may be phenotypically neutral, if flanked by appropriately inherited regions, and suggests that the occurrence of small regions of UPD may be more common than previously suspected. This implies that biparental inheritance should be tested for any time a genetic defect is linked to human disease, especially when there are questions about the recessive or dominant nature of a defect or the associated disorder.