Article Text

Statistics from Altmetric.com
Editor—There have been five previously described cases with a de novo interstitial deletion within the distal long arm of chromosome 17.1-5 We describe a sixth case, 46,XY,del(17)(q22q23.3)de novo. Three patients, including the proband in this report, presented with tracheo-oesophageal fistula (TOF)/oesophageal atresia, highlighting a potential genetic locus for this significant congenital anomaly.
The patient was born at 32 weeks' gestation by Caesarean section for fetal distress and intrauterine growth retardation. The liquor volume was low. He was the first child of a 20 year old mother and 22 year old father who were non-consanguineous and healthy. The birth weight was 1420 g (9th centile) and head circumference was 27 cm (2nd centile). Apgar scores were 7 at one minute and 9 at five minutes. The patient was noted to be dysmorphic at birth and oesophageal atresia was diagnosed at 2 days of age (fig 1). The patient had a small fontanelle, a sloping forehead with wrinkled skin, a round facial appearance, hypertelorism, small eyes with upward slanting palpebral fissures, a broad nasal tip with a short philtrum, a downturned mouth, and thin lips. His palate and ears were normal. He had small nails, proximally placed thumbs, and a deep crease between his first and second toes. He had knee contractures and a fixed flexion deformity of his hips. There was no palpable hip instability and ultrasound confirmed this.

The proband aged 3 days. Note the wrinkled forehead, hypertelorism, small, upward slanting palpebral fissures, broad nasal tip, and downturned mouth. Proximal placement of the thumb is shown on the right.
Echocardiography showed a ventricular septal defect with an overriding aorta, pulmonary valve stenosis, and a left to right shunt. Cranial ultrasound was normal, as was renal ultrasound. At surgery oesophageal atresia was confirmed with tracheo-oesophageal fistulae communicating with both the upper and lower oesophageal pouches. A successful oesophagostomy and gastrostomy were performed. Anal stenosis was noted and required repeated dilatations and eventually an anoplasty was performed.
The baby's recovery was complicated by several episodes of sepsis but he was discharged at 2 months of age. He made little developmental progress. Readmission was required for cyanotic episodes related to a deterioration in his cardiac condition. He developed bronchiolitis and died at 3½ months.
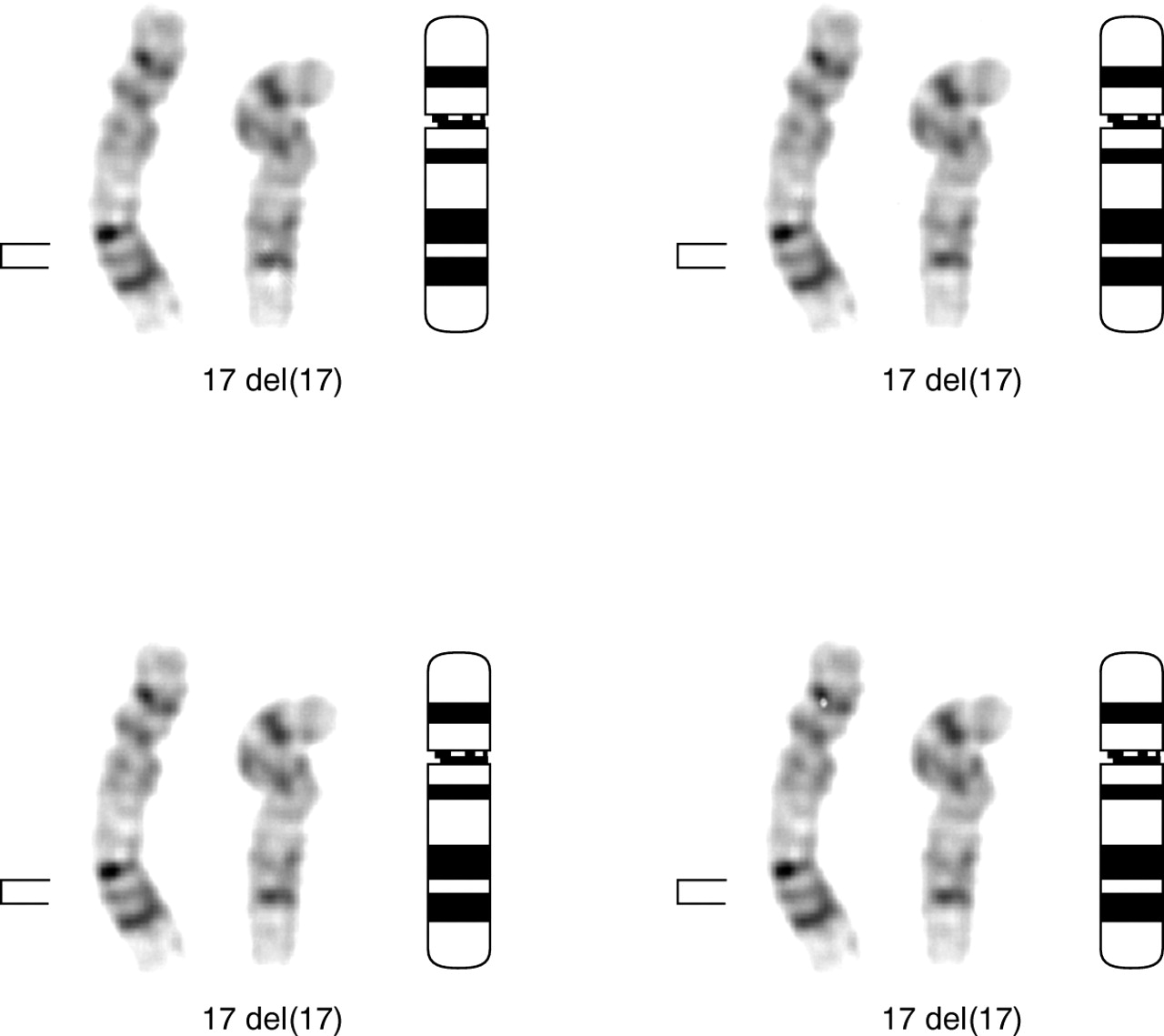
Short term peripheral blood cultures were initiated and harvested by standard protocols. G banded analysis was carried out using trypsin digestion followed by Leishman staining. All metaphases examined showed a male karyotype with an apparent interstitial deletion of the region 17q22→q23.3 (fig 2). Parental karyotypes were normal. The patient's karyotype was therefore 46,XY,del(17)(q22q23.3)de novo.

{kind=link}
{kind=link}
A partial G banded karyotype indicating the normal chromosome 17 (on the left) and the deleted chromosome 17 ( in the centre). A chromosome 17 ideogram is also shown (on the right). The bracket indicates the deleted region.
Five cases of interstitial deletions within the distal long arm of chromosome 17 have been described with a similar phenotype, including microcephaly, facial dysmorphism, proximal placement of the thumbs, and developmental delay.1-5 The features are shown in table1. Congenital heart disease has been reported in all the cases that have died prematurely including the present case and those of Levinet al 4 and Dallapiccola et al.2 The cardiac malformations described in these latter two cases differ from the present case and include bicuspid aortic valve, atrial septal defect, patent ductus arteriosus, and dilated left atrium and ventricle. Both of the other patients have a more distal breakpoint, involving 17q24. The patient in this report has similar breakpoints to that of Mickelson et al,5 who reported a female with no cardiac defects. Our finding is thus consistent with a candidate region for cardiac malformations being more proximal to 17q24 and is against the hypothesis put forward by Mickelson et al,5who suggested a more distal locus for cardiac abnormalities.
Clinical features of the six cases with deletions of chromosome 17(q21-24)
TOF/oesophageal atresia is rarely associated with a consistent chromosome abnormality. Brunner and Winter6 commented that an abnormal karyotype can be detected in 6% of patients with oesophageal atresia and associated anomalies. Schinzel7noted that oesophageal atresia and TOF occur uncommonly in at least 10 different chromosomal abnormalities. These have been isolated cases in all instances except for two, where two out of 15 patients with dup(3)(pter-p21) had a TOF as did two out of three cases with del(6)(q13-q15); there were no cases associated with chromosome 2 abnormalities. The discovery therefore of three patients with an interstitial 17q deletion and TOF is significant and unlikely to be coincidental. The breakpoints seen in our patient (q22-23.3) fall within the same region as reported in the two other patients1 2 and define a possible critical region. This is emphasised by two of the three cases without TOF having more distal breakpoints.4 5 The one case with no TOF and breakpoints in our critical region may confound this hypothesis, but is more likely to be because of variable expression and should not deter further research into this region. There are many other examples of deletion syndromes with a different clinical outcome in the presence of haploinsufficiency to support this.12
It is of note that the clinical findings have some overlap with Feingold syndrome,8 where oesophageal/duodenal atresia is commonly reported along with microcephaly and mesobrachyphalangy. However, the hand and foot findings seen in the 17q deletion cases such as symphalangism and thumb anomalies are not specific for Feingold syndrome and none of the patients have 4,5 toe syndactyly which is characteristic of this inherited condition. A recent report from Celliet al 10 showing linkage to 2p22.3 would confirm these differences.
Human Nog has been shown by Gong et al 11 to map to 17q22. Noggin encodes a bone morphogenetic protein (BMP) antagonist which is expressed in the node, notochord, and dorsal somite. In mice lacking Noggin, cartilage condensations initiated normally but developed hyperplasia. In humans, heterozygous Nog mutations have been shown in multiple synostoses syndrome (SYNS1, OMIM 186500) and proximal symphalangism (SYM1, OMIM 185800). Our patient did not have similar joint abnormalities, nor is TOF a feature of either of these syndromes. Cartilage is, however, present in the oesophagus which could represent a rather tenuous link between these findings.
We believe that this case provides further evidence of a distinct phenotype associated with deletions of 17q22-q23 and confirms an association between haploinsufficiency of this region and oesophageal atresia. This is of particular interest as familial oesophageal atresia is rare9 and there are no other clues to genomic locations of importance for this malformation.
Acknowledgments
We would like to acknowledge the help of the family with this publication.