Article Text

Statistics from Altmetric.com
Editor—Neurofibromatosis type 1 (NF1) is a common human disorder (1/3500 live births) with neuroectodermal involvement resulting in dermatological manifestations of café au lait spots, cutaneous/subcutaneous neurofibromas, and freckling of major folds.1 Hamartomas of the irides (Lisch nodules), well observed on slit lamp examination, are helpful phenotypic markers. Owing to diagnostic uncertainties, especially in young patients, an international scoring system has been discussed and agreed upon.2 Half of the cases result from fresh mutations, others show an autosomal dominant mode of inheritance. The gene mutated in NF1 maps to 17q11.2, is composed of 57 plus at least three alternatively spliced exons,3 and is of ubiquitous expression. The encoded product, referred to as neurofibromin, is a member of the so called GTPase activating proteins (GAPs), and is an upstream downregulator of the p21Ras/Raf/MAPkinase signalling pathway.4 Though genetic homogeneity is a hallmark of this condition, phenotypic heterogeneity has been exemplified by an extreme spectrum of diversity ranging from malformation or malignant variants to virtually benign dermatological changes.1
In particular, and among the many causes for gastrointestinal involvement in NF1 patients, the association with intrinsic intestinal dysmotility (IID), resulting from intestinal neuronal dysplasia type B (IND B)5 6 or aganglionic megacolon (Hirschsprung's disease, HSCR)7 has been documented and is now well established.
We report here a family showing aggregation of NF1 and IID in two sibs, in one of whom congenital megacolon necessitated a Duhamel abdominoperineal pull through with tired suction biopsies of the colon and analysis of the whole excised specimen indicating IND B. This kindred provided a unique opportunity to unravel the genetic bases for the association between two such disorders of neural crest cell development.
The proband was seen at the age of 27 months for investigation of a multiple congenital anomaly/mental retardation (MCA/MR) complex. This young female was born to unrelated white parents. A familial component of both neurofibromatosis type 1 and severe intestinal dysmotility was shown. According to the history, intrauterine growth retardation had been evident from 6 months of gestation. Recurrent caesarean section was performed at 38 weeks of gestation. Birth weight was 2360 g, crown-heel length 46 cm, and OFC 32 cm. The Apgar scores were 8 and 10 at one and five minutes. Congenital heart disease was then diagnosed owing to heart failure with evidence of ventricular septal defect (membranous), persistent ductus arteriosus, and coarctation of the aorta. A two step surgical procedure including tissue grafting led to complete recovery. However, unresolved growth retardation ranging between −3 and −4 SD, cognitive impairment, the presence of multiple cutaneous café au lait spots, and persistent severe constipation indicated a possible MCA/MR syndrome.
On examination, the child was of short stature (76 cm, −3.5 SD), low weight (8.22 kg, −3.25 SD), and had relative macrocephaly (48 cm, ∼mean). A distended abdomen contrasted with a generally wasted appearance (BC/OFC=0.26, normal ⩾0.30). Minor facial anomalies were also noted including frontal bossing, temporal narrowing, depressed nasal root, small, tapered chin, and hypoplastic ear lobes. There were downward slanting palpebral fissures but no true hypertelorism (ICD=25 cm, OCD=67 cm) (fig 1). High vaulted (uncleft) palate as well as hypoplastic and widely spaced teeth were also observed. There was apparent anterior displacement of the anus. The extremities were normal. Dermatological scrutiny showed >10 large (>5 mm) café au lait spots over her trunk and several achromic patches on the four limbs. There were no cutaneous/subcutaneous neurofibromas and axillary freckling was not a feature. The child could not walk unaided but rather crawled on all fours, and speech consisted of only a few words. The developmental quotient score was 57, as ascertained by the Binet test.
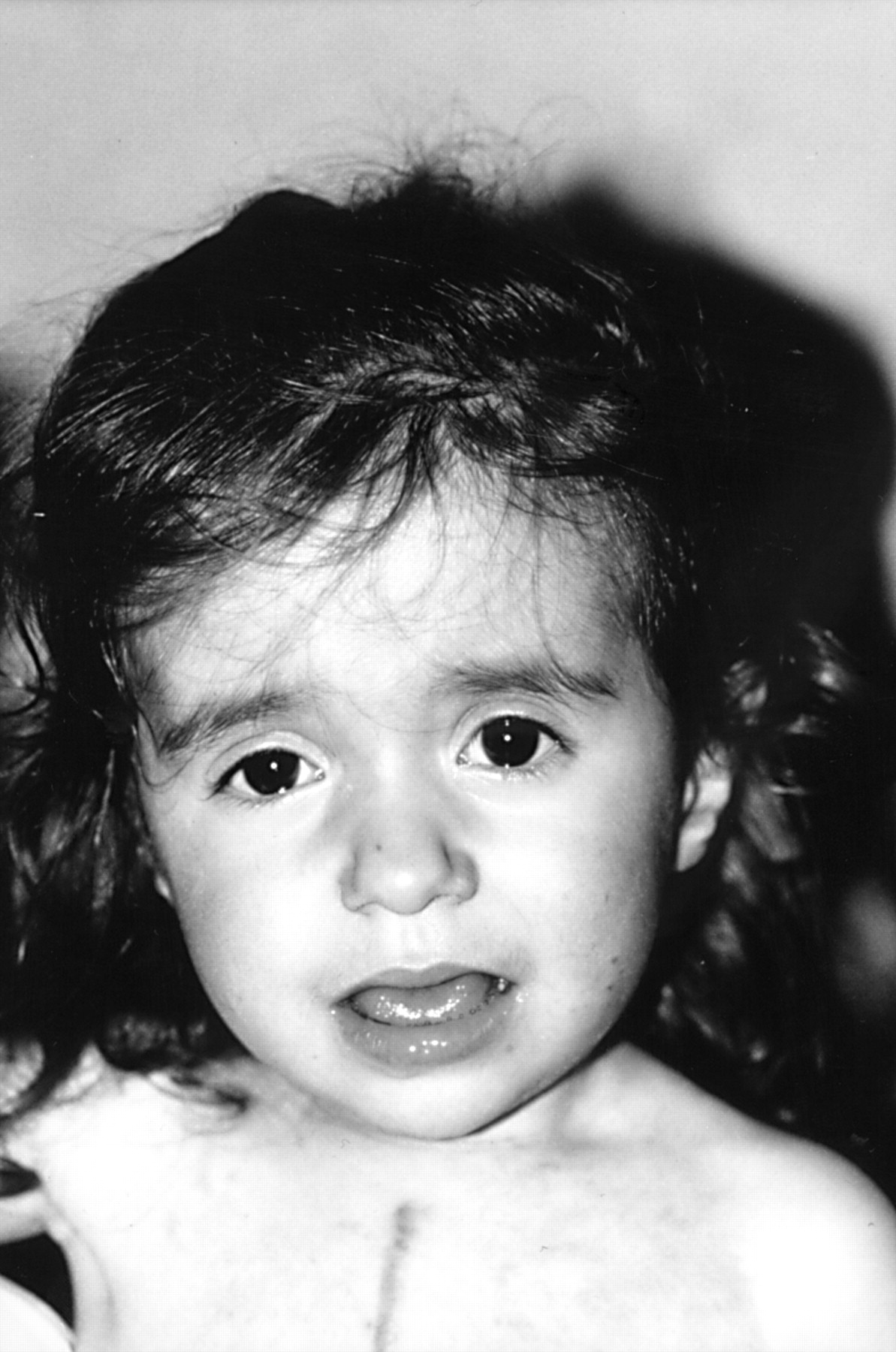
The proband aged 27 months. Note downward slanting palpebral fissures, apparent hypertelorism, depressed nasal root, small, tapered chin, and scar from sternotomy.
Cholesterol was slightly raised (6 mmol/l, normal <5.3), but triglycerides were within normal limits. Metabolic screening was negative. Skeletal x ray showed evidence of delayed bone age (18 months), and soft tissue abdominalx ray showed faecal impaction with stercoliths. Distended intestines on contrast enema indicated megacolon (fig 2). Ophthalmological evaluation, including slit lamp examination of the irides and fundoscopy, was normal. MRI showed enlargement of the right rear aspect of the myelencephalon and a bright signal with contrast enhancement consistent with the diagnosis of hamartoma or low grade glioma. Bright signals were also evident at the medial cerebellar peduncle and in the left semioval centre on T2 weighted sequences, consistent with so called unidentified bright objects (UBOs).

Contrast enema. Distended intestines are consistent with the diagnosis of congenital megacolon.
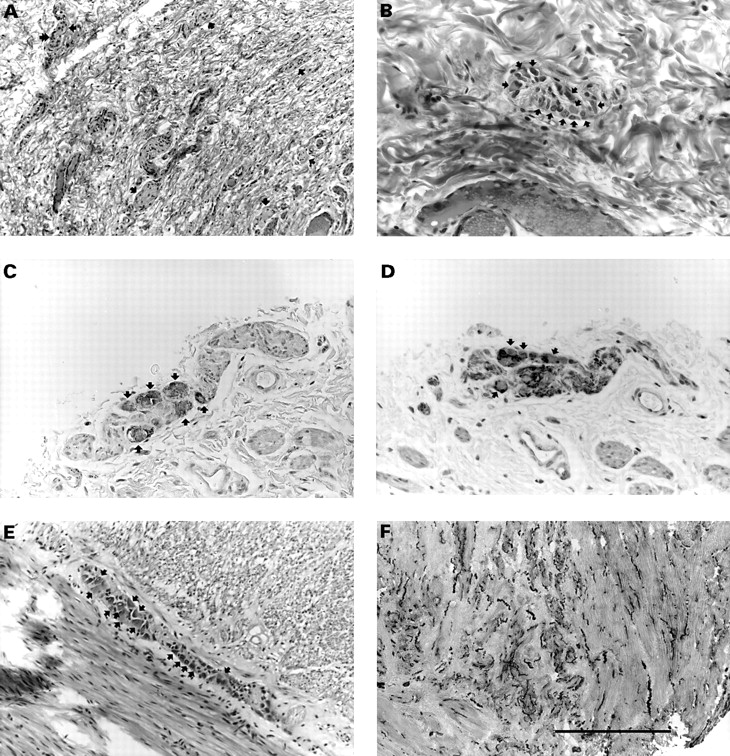
Because of the congenital megacolon, surgical biopsies of the colon and rectum were performed and were found to be consistent with aganglionosis with Schwann cell hyperplasia in the Auerbach plexuses and hypoganglionosis with Schwann cell hyperplasia in the Meissner plexuses. A Duhamel abdominoperineal pull through was thus performed. Analysis of suction biopsies of the rectum, sigmoid, and left colon and of the whole subsequent colectomy specimen pointed to intestinal neuronal dysplasia type B (IND B), based on the presence of abnormal submucosal plexuses showing focal hyperplasia (in terms of density and sizes), occasional giant ganglia harbouring >10 neurones, and nerve cell buds along afferent nerves. The presence of giant ganglia, two to three times as large as their normal counterparts, was set forth as the only reliable, age independent diagnostic criterion for IND B,8 since hyperplasia of the submucosal plexuses, an increase in acetylcholinesterase (AChE) activity in the nerve fibres of the lamina propria, and low SDH activity in nerve cells were shown to normalise with the ongoing maturation of the enteric nervous system. In addition, myenteric plexuses were either normal or hypertrophied, with numerous mature neurones (fig 3).
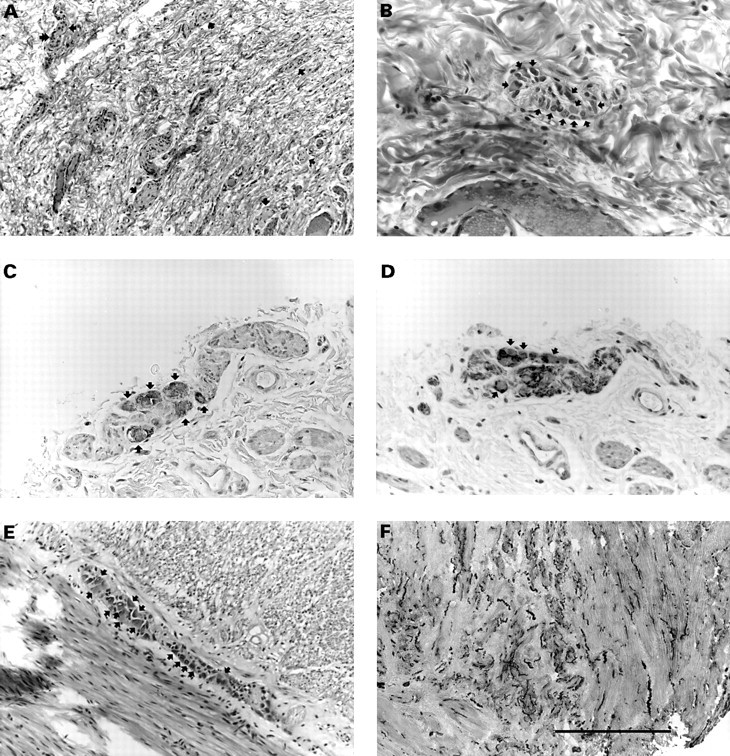
Intestinal neuronal dysplasia. Suction biopsies of the rectum, sigmoid, and left colon, and the subsequent colectomy specimen were routinely processed for histology and immunostained for neurone specific enolase (NSE), a marker of neurones, and protein S100, a marker of Schwann cells, in order to identify intrinsic nervous structures better. In addition, acetylcholinesterase activity could be ascertained from an archival frozen sample. (A) Abnormally increased density of the nervous structures (arrows) and Schwann cell hyperplasia are consistent with hyperplasia of the rectal submucosal plexuses; ganglion cells (double arrow) are occasionally visible at this magnification (haematoxylin, eosin, and saffron, scale=400 μm). (B) “Giant” submucosal sigmoid ganglion, that is, containing more than 10 neurones (arrows) with typical, large, amphophilic cytoplasm (haematoxylin, eosin, and saffron, scale=150 μm). Neurones of a myenteric plexus are visible after immunostaining with anti-NSE antibody (C, arrows) and are negative for antiprotein S100 antibody, which specifically stains the Schwann cells (D, arrows) (serial sections, immunoperoxidase, scale=150 μm). (E) A myenteric sigmoid plexus showing numerous neurones (arrows, haematoxylin, eosin, and saffron, scale=400 μm). (F) Acetylcholinesterase staining of the rectal muscular layer showing numerous, coarse, and undulating fibres (dark) (frozen section, scale=750 μm).
In order to address the possibility of a cytogenetic aberration in relation to a MCA/MR syndrome in the proband, her chromosome complement was established. From analysis of high resolution RBG banded chromosomes, it was apparent that she had a cytogenetically balanced reciprocal translocation, her specific chromosomal constitution being 46,XX, t(15;16)(q26.3;q12.1) (fig 4). This abnormality was inherited from her father (I.1, fig 5A), whose lymphocytes harboured the same translocated chromosomes and was also shared by her older sister (II.1) and healthy brother (II.2) (data not shown; see below for additional family data).

Reciprocal balanced translocation. Wild type as well as derivative chromosomes 15 and 16 from the proband are shown prepared from whole blood lymphocytes with RBG labelling at the 550 band level, following the guidelines of the International System for Human Cytogenetic Nomenclature (ISCN 1995). Arrows point to breakpoints on der(15), in 15q26.3, and on der(16), in 16q12.1.

(A) Family pedigree. Arrow indicates the proband. II.3 was a miscarriage. Complex phenotypic traits are specified as shown in the key. The presence of the reciprocal t(15;16)(q26.3;q12.1) translocation in I.1, II.1, II.2, and II.4 is indicated by an asterisk. The proband and her older sister (II.1) both have NF1 skin symptoms, have severe constipation/megacolon, and harbour the familial cytogenetically balanced reciprocal translocation. (B) Restriction with HphI. The NF1 exon 16 amplimer is shown restricted for I.1 (lane 4), I.2 (lane 5), II.1 (lane 6), II.2 (lane 7), and II.4 (lane 8), co-electrophoresed with molecular weight marker GenLadder TM 100 (Genaxis Biotechnology, Montigny-le-Bretonneux, France; lane 1). Arrowheads indicate theoretical fragment sizes in base pairs (bp) as compared to a healthy control, unrestricted (552 bp fragment, lane 2) and restricted (532 bp fragment, lane 3). Healthy subjects I.1 and II.2 display the expected wild type 532 bp fragment, whereas I.2, II.1 and II.4, diagnosed with NF1, have wild type 532 bp as well as mutant 457 bp and 82 bp fragments. Note the presence of heteroduplexes in lanes 5, 6, and 8.
The mother of the proband (I.2, fig 5A) presented with classical neurofibromatosis type 1 with multiple large café au lait spots and cutaneous/subcutaneous neurofibromas. Her condition was seemingly inherited through the maternal grandmaternal lineage. The father of the proband (I.1) had no apparent dermatological ailment, malformation, or cognitive alteration. The older daughter (II.1) presented with café au lait spots and had some degree of problem with school performance and cognitive impairment (VIQ=76, PIQ=83, TIQ=77, according to WISC-R). She had severe constipation with megacolon and a clinical course typical of HSCR until a favourable outcome followed successful medical treatment. The sibship also included an older brother (II.2) who had not, to our knowledge, been referred for dermatological, morphological, or cognitive problems. A spontaneous abortion, occurring at 2 months of gestation (II.3), was also reported, without further details.
This familial association of NF1 and IND B provided a unique opportunity to test for alternative causal mechanisms, namely the random aggregation of two common monogenic disorders versus a discrete neurocristopathy with possible genotype-phenotype correlation at theNF1 locus or modifying loci lying elsewhere in the genome.
Indeed, although no specific gene for IND B has been found, the latter condition may segregate with typical monogenic HSCR.9Consistently, and following strict diagnostic guidelines, ∼30% of IND B patients have accompanying aganglionosis, that is, HSCR.10 Therefore, screening for a mutation in the genes found to be mutated in monogenic non-syndromic HSCR, that is, theRET proto-oncogene,11-13 the genes encoding endothelin receptor B (EDNRB),9 14 15 or its ligand endothelin 3 (EDN3),16 was a prerequisite to dissecting this familial association further. Molecular analysis of the RET proto-oncogene was carried out using DGGE based on psoralen modified primers for exons 3, 5, 9-13, 16, 18, 20, and SSCP for exons 1, 2, 4, 6-8, 14, 15, 17 and 19, as previously described.13 SSCP in conditions previously determined was similarly carried out for the seven EDNRB 9 and the five EDN3 exons.16 Following these procedures, no mobility shift was observed. We assume that no mutation of the relevant genes was present in the proband's DNA.
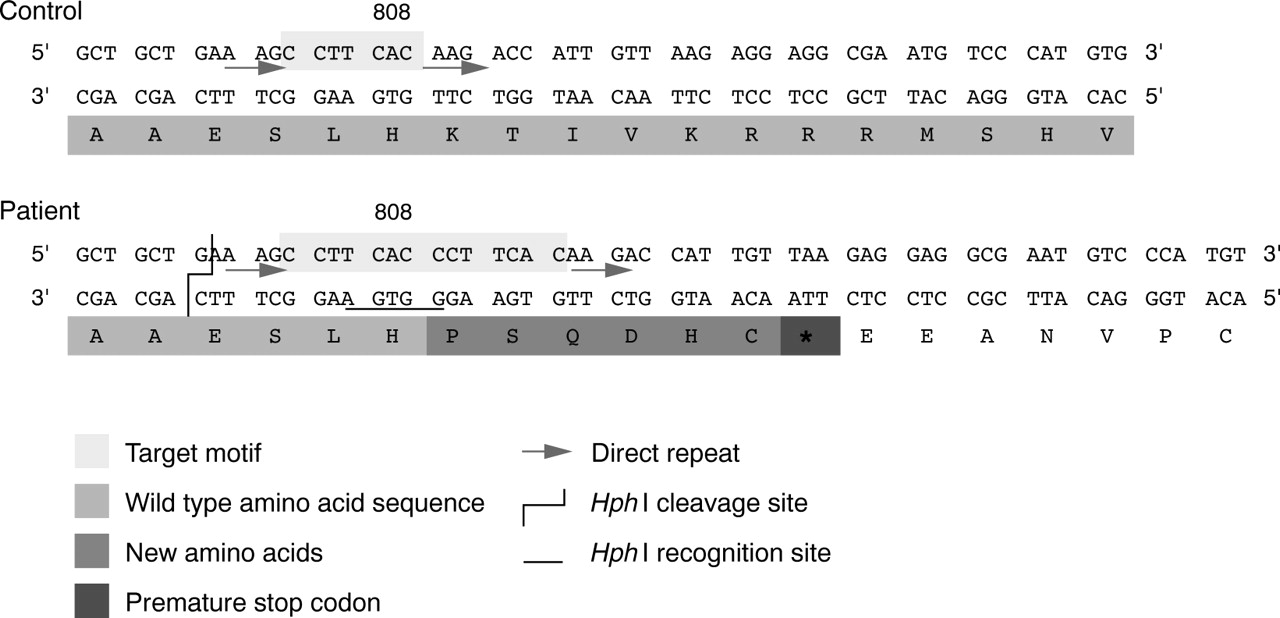
Since none of these genes was faulty in the proband, which favoured a discrete neurocristopathy, it was of particular interest to identify the NF1 lesion. The latter was detected as a result of routine screening for the previously reported recurrent R816X mutation using previously described PCR amplification of NF1 exon 16 followed by restriction with endonuclease HphI.17 Sequence analysis, carried out in conditions previously specified,17 showed a heterozygous tandem duplication of the 7 bp motif (5′ CCTTCAC 3′), nt 2418-2424, inclusive, lying towards the 5′ extremity of the exon and flanked by two short direct tandem repeats (5′ AAG 3′). This DNA lesion (2424-2425insCCTTCAC) caused a frameshift 3′ to codon 808, unchanged, with the addition of six new codons immediately followed by a translation termination signal (TAA), and generated a bona fide recognition site forHphI (5′ GGTGA 3′) (fig 6). This specific lesion featured as a rare mutational event (one case identified among >200 independent NF1 chromosomes tested; data not shown) and was therefore not amenable to genotype-phenotype correlation analysis. However, this out of frame duplication, lying 5′ to the GAP related domain (GRD) specifying region of the gene, was most probably a null mutation and therefore probably did not cause variation in the phenotype.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
NF1 tandem duplication. Double stranded sequence of NF1 exon 16, 5′ extremity, is represented in control and patient with translation into protein. The tandem duplication of a 7 bp target motif flanked by two short direct repeats in the patient results in a frameshift mutation and leads to a putative truncated protein as shown by the insertion of six new amino acids, immediately followed by a premature translation termination codon (TAA). This tandem duplication also generates a recognition site for type II endonuclease HphI, as indicated above.
Using the aforementioned screening method, the status of the rest of the family with regard to the mutation was eventually determined. Predictably, the mother and sister, diagnosed with NF1, were heterozygotes for the 2424-2425insCCTTCAC lesion, whereas the father and older brother were both wild type homozygotes (fig 5B).
Alleles that modify the severity of the phenotype (or penetrance) of a given, well established monogenic factor, that is, mutation ofRET, EDN3, orEDNRB in HSCR, with or without coexisting IND B, have been postulated and some of the relevant genes or loci subsequently identified. The first such example was illustrated by a large inbred Mennonite HSCR pedigree that segregated a missense mutation in EDNRB 18 and otherwise showed linkage disequilibrium with marker alleles mapped to 21q22. This finding was highly suggestive of a HSCR genetic modifier linked to this chromosomal region,19 which might elsewhere account for high HSCR prevalence among trisomy 21 patients.20 More compelling evidence for a HSCR modifier is exemplified by the genes encoding glial cell line derived neurotrophic factor (GDNF)21-23 and, more recently, neurturin (NRTN),24two highly homologous natural ligands of the RET tyrosine kinase receptor protein. Indeed, since GDNF andNRTN were found to be mutated in families also segregating well characterised RETalleles, it was postulated that alterations of these genes were not sufficient in themselves to cause HSCR, but that they probably contributed to the severity of the phenotype, or to higher penetrance of the RET mutations.
As to NF1, analysis of several quantitative or binary traits among a large sample of familial cases concluded that the number of café au lait spots and the number of cutaneous neurofibromas, showing high concordance among MZ twin pairs but weaker concordance in more distant relatives, were determined by non-allelic, “modifying” loci.25 Absence of correlation between the characters analysed suggested that these loci were also trait specific. The presence or absence of intestinal neuronal dysplasia, or bona fide HSCR, is a rare configuration that could not have been assessed. However, modifying loci remain a strong possibility, especially since NF1 and HSCR/IND B are both characterised by disruption or maldevelopment of neural crest cell derivatives. In other words, alleles of genes whose products interfere with the determination, migration, or differentiation of ganglion cells of the enteric plexuses might, in the presence of a perturbed Ras signalling pathway, lead to IID. In this respect, it is noteworthy that the proband's NF1 phenotype is consistent with the involvement of a broad spectrum of neural crest cell derivatives, that is, also including motor neurones of the enteric nervous system, conotruncus, and cranial crest mesectoderm.26
Finally, the most salient aspect of this report is the cosegregation, in two sisters affected with NF1 and megacolon, of anNF1 lesion inherited from the mother, in whom it results in a classical presentation of the condition, and of a cytogenetically balanced reciprocal translocation inherited from the father and shared by an older brother, in whom this chromosomal aberration has no apparent pathogenicity. None of the translocation breakpoints (15q26.3 and 16q12.1) has yet been involved in HSCR or IND B, alone or in a more complex phenotype (see the review by Passarge27 and a recently recognised entity by Mowatet al 28). Therefore, these chromosomal regions provide obvious candidate locations for additional NF1 or HSCR/IND B modifiers or both.
Acknowledgments
The authors are grateful to the patients for their participation, to Dr J-F Chateil for helpful diagnostic information, and to Dr D Récan and co-workers for the establishment and maintenance of lymphoblastoid cell lines. This work was supported by the French Ministère de l'Education Nationale, de la Recherche et de l'Enseignement Supérieur, and the Association pour la Recherche sur le Cancer. MB is the recipient of a scholarship allocated by the Fondation pour la Recherche Médicale.