Article Text

Statistics from Altmetric.com
Editor— Prader-Willi syndrome (PWS)1and Angelman syndrome (AS)2 are clinically distinct neurobehavioural disorders both resulting from altered expression of specific imprinted genes located in the 15q11q13 chromosomal region. PWS is usually caused by a deletion in the paternally inherited chromosome 15 or by maternal uniparental disomy (UPD) of chromosome 15, whereas maternal deletion or paternal UPD is responsible for AS.3 4 AS patients exhibit severe mental retardation with absence of speech, frequent and inappropriate laughter, ataxic gait with raised arms, and a frequent history of seizures. Most patients have a typical face with a wide open mouth, protruding tongue, and prominent chin.5 Clinical history and physical examination are different in patients with PWS, who have neonatal hypotonia almost invariably associated with poor sucking requiring nasogastric feeding, hypogonadism, short stature, mild to moderate mental retardation, and childhood onset obesity owing to hyperphagia beginning between 1 and 6 years.6 However, although all these clinical characteristics are well defined, molecular confirmation is recommended considering that other diseases share identical clinical features, for example fragile X syndrome and PWS,3 7 8 or Rett syndrome and ATR-X syndrome and AS.5 9 The molecular diagnosis of PWS and AS is based on the analysis of the differential parental specific DNA methylation imprint within the 15q11-q13 chromosomal region. This investigation is currently performed by Southern blotting using methyl sensitive restriction enzymes and either probe SNRPN or PW71.10 11 Because most of the PWS and AS patients have a molecular defect of the same chromosomal region, a single molecular test is used for these two different diseases.
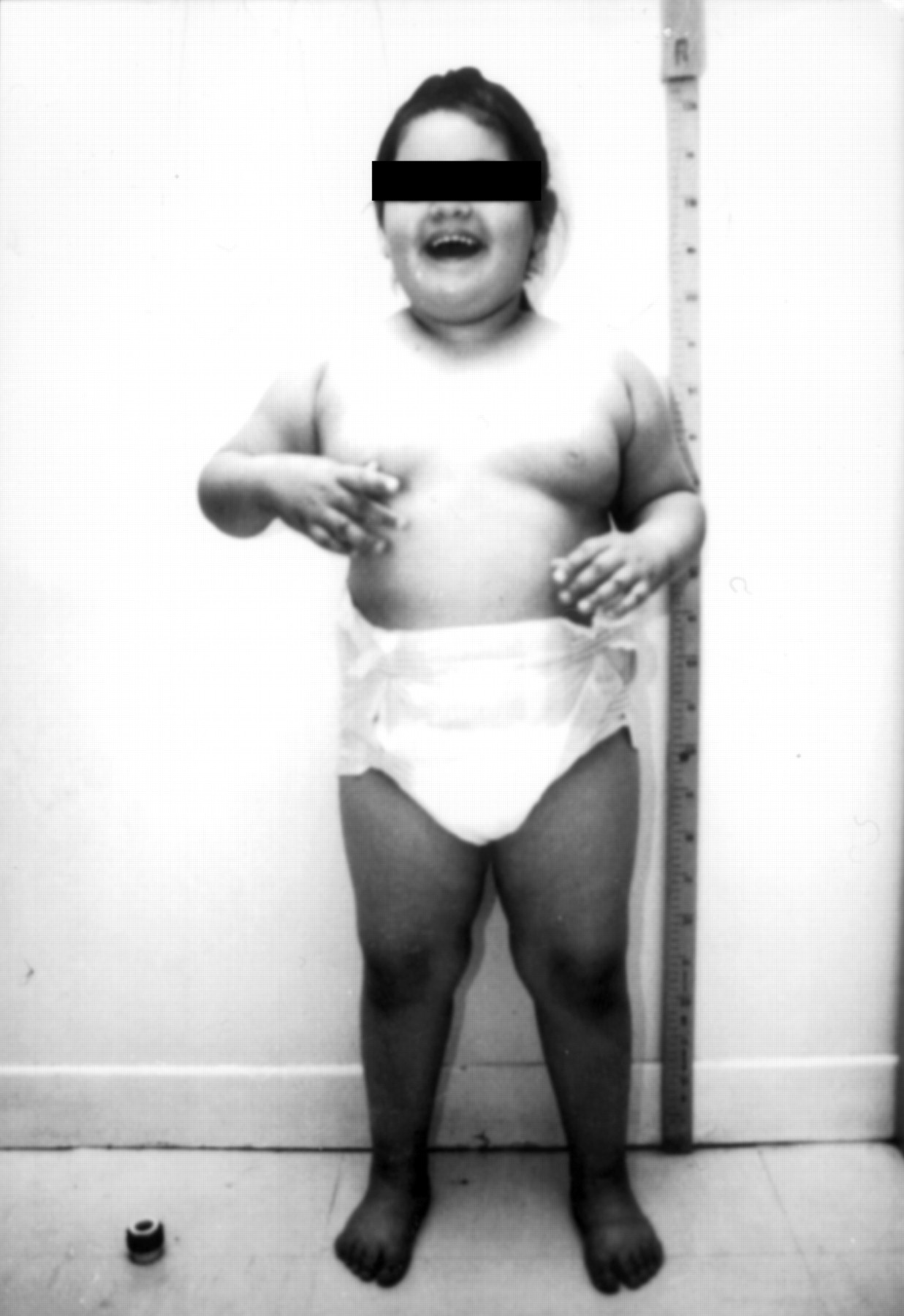
We report on a 5 year old girl born to non-consanguineous, healthy parents, whose clinical history was suggestive of PWS. After an uneventful pregnancy she was delivered by caesarean section because of fetal distress. Apgar scores were 3 at one minute and 6 at five minutes, birth weight was 3040 g, length 50 cm, and head circumference 34 cm. The neonatal period was characterised by hypotonia with feeding difficulties associated with severe gastro-oesophageal reflux. From the age of 2, she became progressively obese as a consequence of hyperphagia. CT scan was normal at 20 months. At 5 years old, physical examination was normal except for the obesity (fig 1). Her weight was 32 kg (>97th centile), length 113 cm (90th centile), head circumference 51 cm (50th centile), and formal developmental assessment showed a developmental quotient of 30 (gait DQ=36, visuomotor coordination DQ=30, socialisation DQ= 25) associated with very poor language skills. No dysmorphic features were observed and she was hyperactive. She has never had any seizures and her gait is not ataxic.

{kind=link}
The proband aged 5 years. (Photograph reproduced with permission.)
DNA methylation testing was carried out to confirm PWS. Genomic DNA was isolated from peripheral blood lymphocytes by conventional methods. Hybridisation of genomic DNA from the proband was performed using probes SNRPN and PW71B, as described previously.10 11 Unexpectedly, both probes showed a single band of paternal origin and no band of maternal origin (data not shown). This pattern showed a complete lack of maternal imprinting as classically observed in AS patients. Consequently, although this patient displays clinical findings of PWS, she has the typical molecular abnormalities of an AS patient.
Analysis of 20 R banded metaphases from the proband showed a mosaic 47,XX,+mar/48,XX,+2mar karyotype, whereas the karyotypes of both parents were normal. Dual colour hybridisation with probes D15Z1 (PCR generated probe)/SNRPN (Oncor®) and D15Z1/D15S10 (Oncor®) showed no 15q11q13 microdeletion. Hybridisation of D15Z1 with the small marker chromosome (SMC) indicated that it originated from chromosome 15. Further characterisation of this SMC showed that it was a small inv dup(15) without the PWS/AS locus. In the absence of deletion and because these markers usually do not have any phenotypic consequences and may not be directly responsible for the clinical manifestations in the proband,12 13 a test for uniparental disomy was performed. Using PCR based analysis, 10 microsatellites markers at the D15S542, D15S543, D15S11, D15S10, GABRB3, GABRA5, ACTC, CYP, FES, and D15S87 loci were analysed by standard semiautomatic methods on an automated ABI PRISM 377 (Perkin Elmer) using fluorescent primers. Of the 10 microsatellites tested, five were fully informative in the family. No maternal allele was observed at D15S542, GABRB3, ACTC, CYP, or D15S87 loci which appeared homozygous for all dinucleotide repeat polymorphisms (table 1). Hence, our proband has inherited two copies of one paternal chromosome 15 (isodisomy) and has a pat UPD(15), as seen in 3-4% of AS cases.14 15
Results of microsatellite analysis. Allele designations are arbitrary
To our knowledge, only two phenotypically atypical AS adults and one child have been reported.16-18 By contrast to our patient, they showed the expected clinical findings of AS with additional PWS features including moderate obesity. Both adults had a cytogenetic deletion of 15q11q13 and the child had a paternal UPD(15). However, our proband lacked the major signs of AS including movement or balance disorder, frequent laughter, inappropriate happiness, and seizures, and only had speech impairment and hyperactivity. The main features typical of PWS were the history of neonatal hypotonia with feeding difficulties and the occurrence of hyperphagia with resulting obesity from the age of 2 years. However, after the molecular results, an EEG was performed that showed the typical slow wave bursts, providing further evidence for a diagnosis of AS. At first sight, the presence of a UPD(15) instead of a deletion does not explain this particular clinical overlap. No striking differences have been reported in AS patients so far19 (although UPD patients may have a slight increase in weight when compared to deletion patients20), and the minor phenotypic differences between UPD and deletion either in PWS or AS patients has never led to a misdiagnosis.21 In a similar way, the finding of the inv dup(15) could not account for this phenomenon as such SMCs do not usually give rise to severe clinical manifestations when they do not include the PWS/AS region.22 However, they are known to be frequently associated with maternal or paternal UPD through different mechanisms.23 In this case the presence of isodisomy rather than heterodisomy is in favour of a postzygotic duplication of the single chromosome 15 in a 46,XX,−15,+inv dup(15) zygote.
The biological basis for this case remains to be resolved, but a simple explanation could be a coincidental association of an incomplete AS phenotype with obesity and neonatal hypotonia. Although clinical PWS and AS overlaps are infrequent events, they nevertheless underline the importance of molecular testing in the diagnosis of these syndromes.3 The study of such atypical cases will probably contribute to a better understand of their physiopathology.
Acknowledgments
Drs Dupont and Cuisset contributed equally to this work.