Article Text

Statistics from Altmetric.com
- BWS, Beckwith-Wiedemann syndrome
- DMR, differential methylated region
- OGS, overgrowth syndromes
- SGBS, Simpson-Golabi-Behmel syndrome
- overgrowth syndrome
- mannose 6 phosphate/insulin-like growth factor 2 receptor
- parental imprinting
- IGF-II, methylation
Overgrowth syndromes (OGS) comprise different disorders with overlapping phenotypes. They demonstrate a variety of features, including pre- and post-natal overgrowth, macroglossia, organomegaly, abdominal wall defects, and hypoglycaemia. They also predispose to the development of embryonic tumours (most commonly Wilms’ tumour).1 The pathogenesis of two different OGS is well established. Beckwith-Wiedemann syndrome (BWS; OMIM #130650) is related to genetic or epigenetic changes in the imprinted 11p15 region resulting in an increased level of IGF2 (reviewed by Maher & Reik2). Simpson-Golabi-Behmel syndrome (SGBS; OMIM #312870) is an X linked disease attributed to mutations in the glypican-3 gene (GPC3), which encodes an extracellular proteoglycan believed to interact with insulin-like growth factor II (IGF2) 3,4 and/or other growth factors.5,6
The proliferative effects of IGF2 are mediated by the type 1 IGF receptor. In contrast, the mannose-6-phosphate/IGF2 receptor (IGF2R) has an anti-proliferative function, by ensuring the clearance and inactivation of IGF2.7 This function of IGF2R is well supported by knockout mouse models; mice lacking functional IGF2R (KO-IGF2R) are up to 30% larger than their wild type littermates.8–10 The increased growth of KO-IGF2R mice can be attributed to higher IGF2 levels because this phenotype is corrected in KO-IGF2R mice that also lack functional IGF2 or IGF1R.9,10
IGF2R is developmentally regulated and its expression is maximal during fetal development and organogenesis.11 Its expression pattern correlates with that of the IGF2 and GPC3 genes.12–14 In the mouse, IGF2R is subject to parental imprinting with a maternal specific expression, and this pattern is maintained throughout development and in all somatic tissues in the adult.15,16 In humans, the imprinting status of IGF2R is controversial. Some data are supportive of a polymorphic imprinting that occurs in the pre-term post-implantation embryo in 25–50% of individuals17–19 (reviewed by Wutz et al20 and Wutz & Barlow21). However, other data suggest that IGF2R is not imprinted in primates, including humans.22
Human and mouse IGF2R genes contain two differential methylated regions (DMRs). DMR1 in the IGF2R promoter is differentially methylated only in mice whereas DMR2, in intron 2, is specifically methylated on the maternal allele in both mice and humans.23–25 DMR2 acts as a promoter for an antisense RNA that is also imprinted in mice, with expression from the paternal allele.26,27 The function of the antisense RNA is not completely understood but the deletion of the paternal DMR2 restores biallelic expression of the sense IGF2R transcript, consistent with an expression competition model.26
In humans, IGF2R has been implicated as a tumour suppressor gene in various human tumours such as breast28,29 and hepatocellular carcinomas30–32 and choriocarcinomas.33,34
Key points
-
Overgrowth syndromes (OGS) in humans include the extensively studied Beckwith-Wiedemann syndrome (BWS) and Simpson-Golabi-Behmel syndrome (SGBS), which have overlapping phenotypes. The most frequent, BWS, results from various genetic or epigenetic modifications of imprinted genes in the 11p15 chromosomal region. SGBS is an X linked syndrome involving mutation in the glypican-3 gene, which encodes a heparan sulfate proteoglycan. In both syndromes, the crucial genetic defect is responsible for an increased availability in insulin-like growth factor II (IGF2) or other growth factors.
-
The mannose-6-phosphate/IGF2 receptor (IGF2R) has an anti-proliferative function and ensures the clearance and inactivation of IGF2. The IGF2R gene is imprinted in the mouse and its disruption results in overgrowth in mice. Furthermore, epigenetic modifications of IGF2R lead to fetal overgrowth in sheep.
-
The aim of this study was to evaluate the role of IGF2R in OGS. Among a series of 152 patients referred to our department for OGS, 55 (36.2%; 30 males and 25 females) did not exhibit abnormalities at the 11p15 locus (BWS) and were included in this study. We analysed the expression of IGF2R and the methylation status of two differential methylated regions (DMRs) of the IGF2R gene in this series of 55 OGS patients (30% with a severe form and 70% with a mild form). One in 55 patients exhibited a partial demethylation of DMR2, indicative of a mosaicism. This pattern was found in different tissues (blood, skin fibroblasts, and tonsils). The partial demethylation of DMR2 was associated with a slight decrease in the soluble form of IGF2R in plasma and with a decrease in IGF2 binding.
-
We show here that although IGF2R is not frequently involved in human OGS, it may play a role in a subset of cases.
Recently, it has been shown that pre-implantation embryo procedures in sheep may result in an inappropriate epigenetic modification in the imprinted IGF2R gene during early embryogenesis and the demethylation of DMR2, leading to downregulation of the receptor and consequently to OGS.35
We and others have described that about 70% of patients referred with OGS exhibit abnormalities at the 11p15 imprinted locus, mostly epigenetic modifications.36–40 Other mechanisms may induce an overgrowth syndrome by inducing the activity of growth factors such as IGF2. Indeed, we have also shown that about 10% of male patients without abnormalities in the 11p15 region exhibited mutation in GPC3 (unpublished data). Alternatively, other IGF2 related regulatory genes, such as IGF2R, are also good candidates for causing OGS.
The principal aim of this study was to evaluate the role of IGF2R in OGS by analysing the expression and the methylation status of the IGF2R gene in a series of OGS patients with no abnormalities in the 11p15 region or GPC3. We show here that although IGF2R is not frequently involved in human OGS, it may play a role in a subset of cases.
MATERIALS AND METHODS
Patients
From a series of 152 patients referred to our department for OGS, 92 (60.5%) exhibited an abnormality at the 11p15 locus. Most had an epigenetic defect: isolated demethylation of KvDMR1 (n = 52) or isolated hypermethylation of the H19 gene (n = 13). Twenty-five patients exhibited a genetic defect, 11p15 uniparental disomy (n = 22) and germline CDKN1C mutation (n = 3). Two patients had an 11p chromosomal abnormality. GPC3 mutations were found in five cases.
The 55 (36.2%) remaining patients (30 males and 25 females) were included in this study. Informed consent for access to the collected information was obtained from all patients (or their parents) in accordance with national ethics regulations.
Patients were assigned to two groups according to their phenotype. Group 1: severe form of OGS (n = 16). These patients presented with at least three of the following four major signs: macrosomia, macroglossia, organomegaly, or abdominal wall defect associated with other clinical features such as neonatal hypoglycaemia, ear abnormalities, or facial naevus flammeus. Four patients had heart disease. Group 2: mild form of OGS (n = 39). These patients presented with less than three of the four major signs. Six had heart disease.
Inheritance
Two of the 55 patients had a monozygotic twin who also exhibited an OGS. One female patient had three brothers and one sister with OGS.
Control subjects
Plasma and DNA samples from, respectively, 57 (55 children) and 25 healthy subjects were used as controls.
Methods
DNA analysis
DNA was extracted from leukocytes (all patients) and tissues (n = 7: tongue tissue obtained from tongue resection for macroglossia (n = 3), normal kidney (n = 1), peritumoural liver (n = 1), and tumours (n = 2)), as previously described.41 Parental leukocyte DNA was available for 51% of the patients.
Allele specific methylation of DMR1 and DMR2 of the IGF2R gene
DMR1 and DMR2 methylation status was assessed by Southern blotting and by methylation specific PCR after bisulphite treatment.
For the Southern blot, briefly, genomic DNA was digested with HindIII and then with the methylation sensitive enzyme HpaII or its non-methylation sensitive isochizomer MspI. Digested samples were subjected to electrophoresis in 1% agarose gels, blotted onto GeneScreen Plus membranes (NEN Life Science Products, Boston, MA, USA), and hybridised with DMR1 or DMR2 probes.
The DMR1 and the DMR2 probes were obtained by PCR. The 5′ sense primer DMR1S (5′-CAGCTGGCCTAGTGTGGTTGAA-3′), and the 3′ antisense primer DMR1AS (5′-TGGCAGATTTGTCATCCTAAGA-3′) amplify a 400 bp fragment in the DMR1 region (GenBank accession no. AF348209, nt 2016–2415). The 5′ sense primer DMR2S (5′-CATGCGCCTCCAGCTGCGCATCTC-3′) and the 3′ antisense primer DMR2AS (5′-GCTGATCCAATCCCATATACACCT-3′) amplify a 410 bp fragment in the DMR2 region (GenBank accession no. X83701, nt 3012–3419).
For the combined bisulphite restriction analysis of DMR2, briefly, after a first step of alkaline denaturation, genomic DNA (1 µg) was deaminated by sodium bisulphite overnight at 55°C. DNA was desalted using the Wizard Clean-up system (Promega, Madison, WI, USA), desulphonated for 30 min at 37°C (0.3 mol/l NaOH) and precipitated.42 The bisulphite treated DNA was PCR amplified with the 5′ sense primer 5′-GGTATGTTGGGGATAGGTTTTGGGAGTTG-3′ and the γP32 labelled 3′ antisense primer 5′-CAAACACACTAACAACCACTACATCCCTC-3′. PCR products were then digested with the methylation sensitive enzyme BstUI. After treatment by bisulphite, the demethylated allele (217 bp) loses this restriction site and is not cut by BstUI. The digested samples were subjected to electrophoresis in SDS/10% PAGE.
IGF1, IGF2, soluble IGF2R assays in plasma
Plasma samples were available for 16 patients (eight patients from each group) and 55 control children. The soluble form of IGF2R (sIGF2R) was determined by ELISA as previously described.43 IGF2 and IGF1 were assessed by RIA as previously described.44,45
Superose-12 gel chromatography, IGF2 binding assay, and affinity labelling of IGF2R
Plasma samples (200 µl) were loaded onto a Superose-12 gel permeation column (Pharmacia Biotech, Uppsala, Sweden) eluting at 1 ml/min in PBS. Fractions (0.5 ml) were collected and the column was washed between runs. The elution of the sIGF2R in fractions 19–21 was confirmed by ELISA, Western immunoblotting and by IGF2 affinity labelling.
The IGF2 binding assay was performed by incubating 150 µl of fractions 12 to 34 in the presence of 125I IGF2 (8000–10 000 cpm) in 25 mmol/l Hepes pH 7.4, 0.1% BSA, and 0.1% Triton X-100. After an overnight incubation at 4°C, IGF2 binding complexes were precipitated by centrifugation after addition of 10 µl 4% gammaglobulin (Sigma, St Louis, MO, USA) and 1 ml of 18% PEG 6K. The pellet was counted in a gamma counter.
Affinity labelling of IGF2R: 20 µl of pooled fractions 19–21 were incubated with 125I IGF2 (100 000 cpm) for 16 hours at 4°C and the samples were then incubated with 0.5 mmol/l BS3 (Pierce, Rockford, USA) for 30 minutes at room temperature. The reaction was stopped with Laemmli buffer and 4% β mercaptoethanol, and samples were submitted to SDS/6% PAGE. The gel was stained and dried before autoradiography.
Densitometry and calculation of the methylation index of DMR2
Blots were analysed with a Storm PhosphorImager (Molecular Dynamics, Inc, Sunnyvale, CA, USA). The methylation index of DMR2 was assessed by densitometry of autoradiographs and calculated as the ratio of the intensity of the methylated band to the sum of the methylated and unmethylated band intensities.
RESULTS
DMR1 and DMR2 methylation analysis in blood samples and tissues
DMR1 methylation analysis was performed in 25 control subjects and 55 OGS patients. Southern blot analysis showed only one band of 2.6 kb after digestion with HindIII and HpaII, confirming that the maternal and the paternal alleles were not differentially methylated25 (fig 1A).

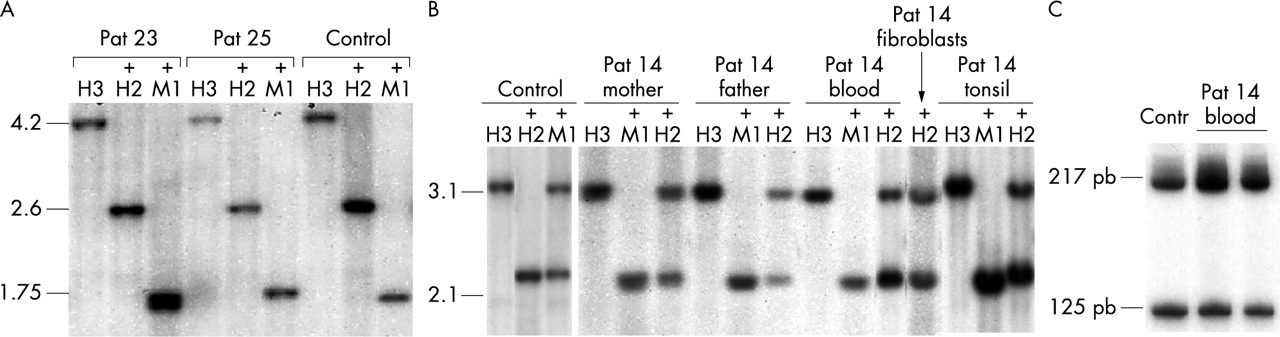
Methylation analysis of DMR1 and DMR2 of IGF2R in patients with overgrowth syndromes. (A) DMR1 methylation was assessed by Southern blotting after digestion with HindIII (H3), HindIII+HpaII (+H2) and HindIII+MspI (+M1). The 4.2 kb HindIII fragment is fully cut with HpaII resulting in a 2.6 kb fragment, indicating that DMR1 is fully demethylated. (B) DMR2 methylation was assessed by Southern blotting after digestion with HindIII (H3), HindIII+HpaII (+H2) and HindIII+MspI (+M1) of leukocyte DNA from a control subject, patient 14, and the parents of patient 14 (as well as skin fibroblasts and tonsil from patient 14). DMR2 is partially demethylated in the different samples from patient 14. (C) DMR2 methylation was assessed by combined bisuphite restriction analysis. After bisulphite treatment, DNA was amplified by PCR (see methods) and digested with the methylation sensitive enzyme BstUI. The 217 bp fragment is the demethylated fragment and the 125 bp fragment is the methylated fragment.
For DMR2, Southern blot analysis of leukocyte DNA from control subjects showed two bands (2.1 and 3.1 kb) of equal intensity after digestion with HindIII and HpaII, indicating that the two parental alleles were differentially methylated (fig 1B). DMR2 has been previously shown to be hypermethylated on the maternal allele.25 DMR2 methylation index was normal, at between 46.6 and 52.6% (mean (SD) 49.5 (1.8)%).
The pattern of DMR2 methylation in leukocyte DNA was normal for 54 of 55 OGS patients with methylation indexes varying between 46.3 and 53.3% (mean (SD) 49.8 (1.5)%). This pattern was also normal in the seven tissues analysed.
One of the 55 patients (patient 14, group 2) exhibited a partial demethylation of DMR2 in blood cells. Skin fibroblasts and tonsil tissues were also available for this patient and the demethylation pattern was the same as in blood cells, with methylation indexes of 31%, 40%, 32%, and 36% in respectively blood cells, skin fibroblasts, and left and right tonsils (fig 1B). Both parents had normal methylation patterns (fig 1B). Combined bisulphite restriction analysis confirmed this partial demethylation pattern (fig 1C).
Analysis of the soluble form of IGF2R (sIGF2R) in plasma samples
Plasma levels of sIGF2R were assessed in 57 (55 children) control subjects and 16 (15 children) patients with OGS (fig 2A). Most patients had levels in the normal range for age, although three had levels below this. Patient 14 had a decreased sIGF2R level (0.57 µg/ml; normal for age 1.12 (0.28) µg/ml) compared with patients of the same age (3 years). However, IGF1 and IGF2 plasma levels were in the normal range (figs 2B and 2C). As in control subjects, IGF1 levels in OGS patients increased with age through puberty.

sIGF2R (A), IGF2 (B), and IGF1 (C) levels in normal subjects (white circles) and patients with OGS (black circles). Patient 14, with partial demethylation of DMR2, is indicated by a grey star.
IGF2 binding assay after Superose-12 gel chromatography
Superose-12 gel chromatography was performed for 13 patients (six patients from group 1 and seven patients from group 2) and compared with samples from age matched control subjects.
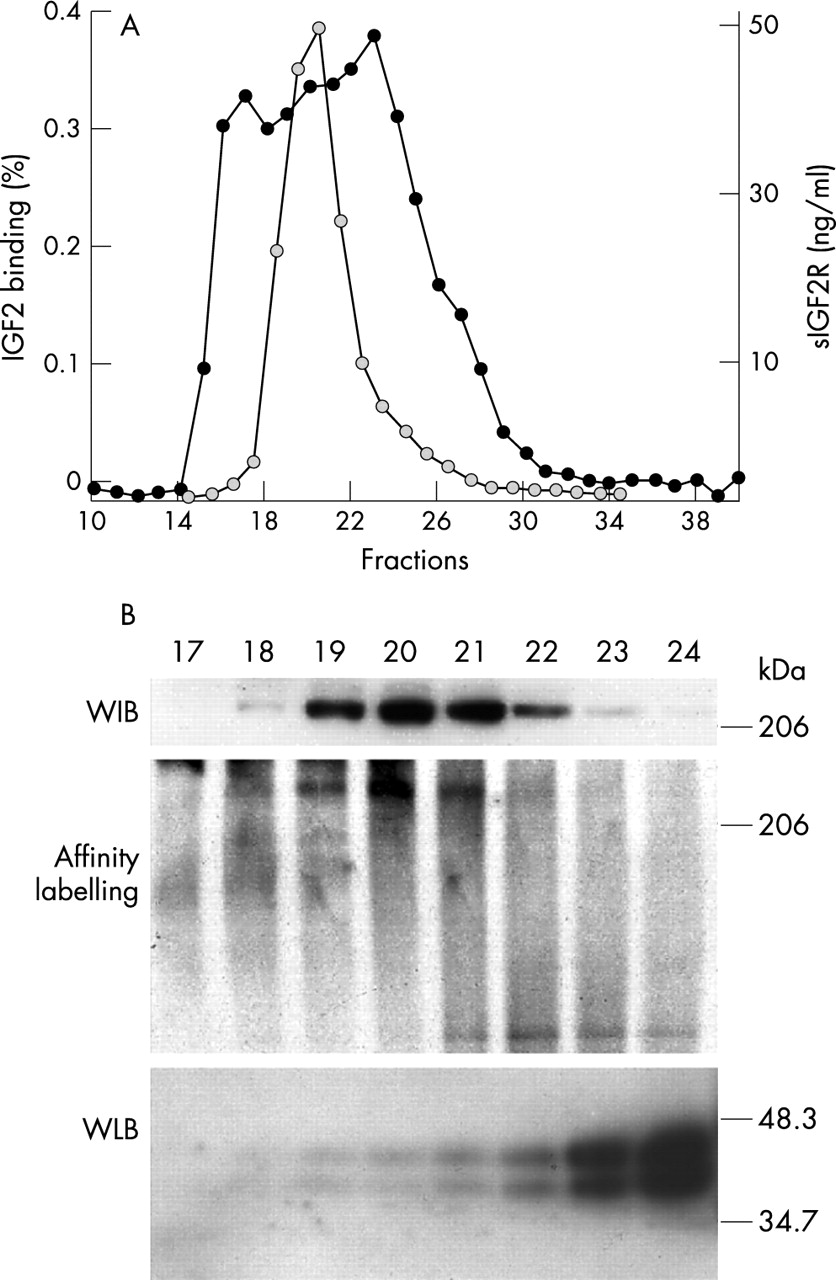
IGF2 binding assay after superose-12 gel chromatography of plasma from control subjects showed three binding peaks. sIGF2R elutes in fractions 19–22 as shown by ELISA (fig 3A), Western immunoblotting, and IGF2 affinity labelling (fig 3B). The IGF2 binding activity eluting in fractions 23–28 correspond to IGFBPs as previously reported46 and shown by Western ligand blotting (fig 3B).

(A) IGF2 binding assay (black symbols) and sIGF2R levels (grey symbols) in Superose-12 gel chromatography fractions of plasma from a normal subject. (B) Western immunoblotting with a polyclonal anti-IGF2 receptor antibody, IGF2 affinity labelling, and Western ligand blotting of Superose-12 gel chromatography fractions of plasma from a normal subject.
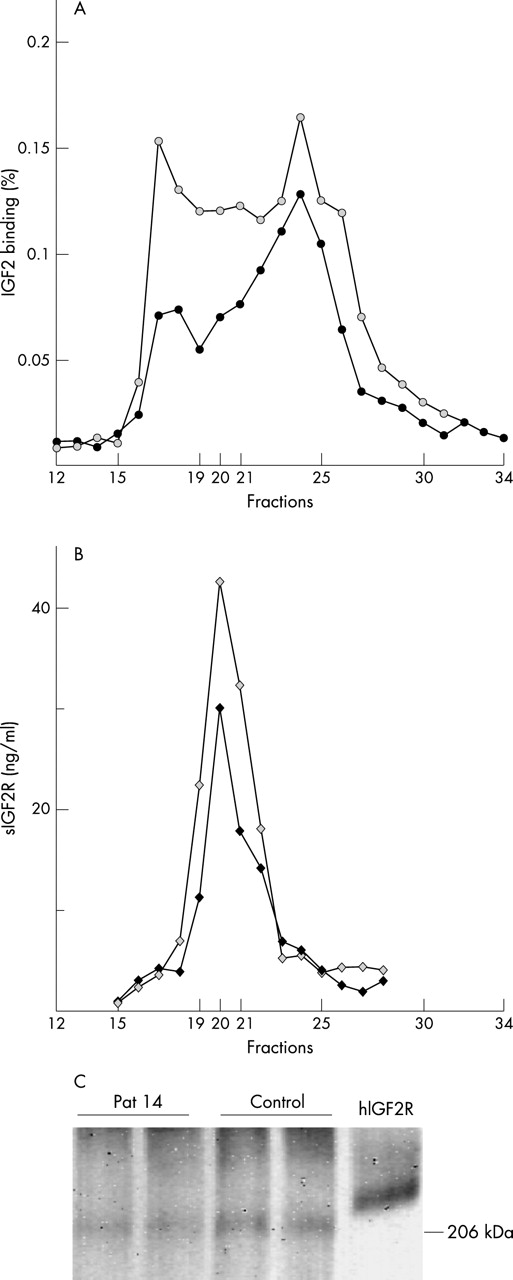
Patient 14, with partial demethylation of DMR2, exhibited a 40% decrease in IGF2 binding of fractions 19–21 when compared with control subjects (three different control subjects (fig 4A). This decrease in IGF2 binding was reflected by a similar decrease in sIGF2R levels (fig 4B). Affinity labelling of IGF2R in pooled fractions 19–21 with125I IGF2 was decreased to a similar extent (fig 4C).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
IGF2 binding assay (A) and sIGF2R levels (B) after Superose-12 gel chromatography of plasma from patient 14 (black symbols) and a representative age matched control subject (grey symbols). (C) Affinity labelling of IGF2R of pooled fractions 19–21 incubated with 125I IGF2 (in duplicates) after Superose-12 gel chromatography of plasma from patient 14 and a representative age matched control subject. The hIGF2R used as a control is a purified membrane receptor and is therefore larger than truncated circulating protein.
DISCUSSION
Experiments in mice and additional findings in humans have increasingly linked the IGF system, and more particularly IGF2, directly to OGS. IGF2 levels are increased and mice are larger when the paternally imprinted H19 gene is deleted47 or when IGF2R is disrupted.8–10 These effects are very probably attributable to excessive IGF2 function, because a further knockout of IGF2 in IGF2R or H19 deficient animals rescues the phenotype.9,10,47 Perhaps more striking is the production of a set of phenotypes quite reminiscent of SGBS or BWS in transgenic mice with an additional copy of IGF2.48
OGS such as BWS and SGBS exhibit overlapping phenotypes, and molecular analysis has already led to diagnosis of SGBS in patients initially considered as BWS. In both syndromes, the fetal overgrowth is, at least partially, related to increased availability in IGF2. Molecular abnormalities of the 11p15 imprinted region or the GPC3 gene account for about 75–80% of patients referred for OGS.
The crucial role of IGF2 for fetal growth and the function of IGF2R in the clearance of IGF2 justified a study of the putative involvement of IGF2R in patients without genetic or epigenetic abnormalities in the 11p15 region. This hypothesis was recently strengthened by the description of epigenetic abnormalities of IGF2R resulting in abrogation of IGF2R expression in cloned sheep with large offspring syndrome.35
IGF2R imprinting is still controversial in humans. IGF2R was initially reported to be expressed from both parental alleles.49,50 However, subsequent studies brought evidence that IGF2R is a polymorphic trait in humans and that IGF2R is imprinted during early development in 25–50% of fetuses.17,18 More recently, Oudejans et al19 confirmed IGF2R imprinting in 50% of first trimester placental tissues. In contrast to the mouse,51 they failed to detect AIR RNA (antisense transcript) in human tissues, even in samples that downregulate IGF2R expression in an allele specific manner. However, a more recent study, analysing different species, reported that in contrast to most imprinted genes, the imprinting of IGF2R is not conserved between mammals and that virtually all primate mammals possess two activated copies of IGF2R.22
Until recently, the human DMR2 was considered as fulfilling the criteria for an imprinting box. However, Killian et al22 also showed that the existence of a DMR in intron 2 is not essential for the imprinting of IGF2R (marsupial opossum) and that a DMR in intron 2 can exist independently of the imprinting of IGF2R (human).
Abnormal imprinting of IGF2R has been reported only once in humans. Xu et al52 showed that 50% of Wilms’ tumours, a very frequent tumour in OGS, exhibited an abnormal imprinting pattern of IGF2R with marked repression of the paternal allele. However, these data were not confirmed by Killian et al.22
The data shown here do not support the hypothesis that the downregulation of IGF2R is a frequent cause of OGS in humans. Indeed, from 55 patients with OGS and with a normal analysis at the 11p15 locus, only one patient exhibited abnormalities of IGF2R both at the DNA and protein levels. The phenotype of this patient was mild with moderate statural advance, macroglossia, and hemihyperplasia. It was very different from phenotypes exhibited by IGF2R deficient mice or by cloned sheep but, in these two models, the defect was responsible for a complete abrogation of IGF2R. Indeed, when reported in sheep,35 the demethylation of DMR2 was complete. In contrast, patient 14 exhibited a partial demethylation of DMR2 that was found in different tissues. The fact that the demethylation of DMR2 was partial is not surprising. In a study of demethylation of the KCNQ1OT gene (11p15 locus) in BWS, we previously showed that at least one third of patients exhibited a partial demethylation, suggesting that this abnormality can occur as a mosaicism with variable tissue distribution.38 The mosaic distribution may also explain the hemihyperplasia in patient 14. The partial demethylation of DMR2 in patient 14 was shown with two different techniques (Southern blotting and combined bisulphite restriction analysis) and in three different tissues. It is noteworthy that this pattern was not more obvious in skin fibroblasts taken from the side of the body with hemihyperplasia.
When the expression and the functionality of IGF2R were analysed, results were concordant with the extent of the epigenetic abnormality. Indeed, the soluble form of IGF2R was slightly decreased in plasma and its binding capacity (analysed by binding assay and affinity labelling) was decreased to the same extent.
In summary, epigenetic modifications of IGF2R are not frequently involved in human OGS, but might play a role in a subset of cases. Other mechanisms of inactivation of IGF2R, such as genetic mutation, are unlikely, as most children with OGS have a normal sIGF2R level. Other mechanisms may cause an OGS by inducing the activity of growth factors and further studies of 11p15 genes and other IGF2 related regulators should be carried out to validate this hypothesis.
Acknowledgments
This work was supported by the Assistance Publique Hôpitaux de Paris, the University Paris VI and INSERM (U515).