Article Text

Statistics from Altmetric.com
Rett syndrome (RS, MIM 312750) is an X linked dominant neurodevelopmental disorder, which has been considered to affect girls only.1 Males were thought to be aborted spontaneously or to have a different phenotype.2
The disease is caused by mutations in MECP2, encoding a methyl-CpG binding protein MeCP2.3 MeCP2 is an abundantly expressed protein acting as a global transcription repressor.4 The protein contains two domains, the 85 amino acid methyl-CpG binding domain (MBD) and the 102 amino acid transcriptional repression domain (TRD).4,5 Furthermore, a domain in the C terminal part of the protein, facilitating DNA binding and harbouring the nuclear localising signal (NLS), has been described.6
The mutation responsible for RS has now been found in the majority of RS patients and more than 100 different mutations have been published. Although some association between phenotype and genotype has been shown, their correlation is not predictive of the clinical manifestations in the individual case.7,8
Since 1999, when mutations in MECP2 were first reported in patients with RS,3 it has been possible to analyse boys for mutations resulting in a confirmation of the diagnosis in some of the earlier reported cases.9,10
Screening of groups of patients with mental retardation of different types has further identified MECP2 mutations in males. So far, 17 cases of males with a disease causing mutation in MECP2 have been found. However, six of them were found to have Klinefelter syndrome or were mosaics for the mutation in question.8–15 The latter can roughly be divided into two groups, the severe neonatal cases, who die within the first years of life, all caused by an MECP2 mutation that in females causes classical RS,8–10 and the non-specific mental retardation type with mutations that, if present in females, is compatible with normal or mildly retarded development.16–19
Here we report an 11 year old boy with classical RS, a normal karyotype, no signs of mosaicism, and a de novo truncating mutation (816dup7) in the MECP2 region encoding the transcriptional repression domain, TRD, in MeCP2.
CASE REPORT
The patient was born at term after a normal pregnancy as the only child of a 24 year old woman and a non-consanguineous, 36 year old man. There are two healthy paternal half sisters. The delivery was by vacuum extraction because of signs of asphyxia. Apgar scores were 7 at one minute and 9 at five minutes, birth weight 2950 g, length 50 cm, and head circumference 37 cm.
He was breast fed for the first months, but was slow to suck. After the shift to formula feeds, he vomited profusely daily and from then on suffered from failure to thrive and retarded growth.
Contact and motor development were considered normal for the first 6 to 7 months. However, he did not sit unsupported before 10 months of age and he never crawled. He used his hands for finger feeding and was able to hold a spoon. He babbled at 5–6 months of age, but never spoke words. Around 1 year of age he lost his babbling sounds and the ability to feed himself, and at 15 months of age he was generally retarded. He has never had the typical stereotypic hand movements, but around 2 years of age he started rubbing his eyes stereotypically.
Key points
-
Rett syndrome (RS) used to be considered as affecting girls only. However, 17 males with a mutation in MECP2 have been reported so far. Six of these had the karyotype 47,XXY or were mosaics for the mutation. The remaining cases can be divided into two groups, a severe type, who die within the first years of life, all caused by an MECP2 mutation that in females has been found to be associated with classical RS, and a mental retardation type with mutations that, if present in females, are compatible with a normal or mildly retarded development.
-
Here we report an 11 year old boy with classical RS, with a normal karyotype, no signs of mosaicism, and a truncating mutation (816dup7) in the MECP2 region encoding the transcription repression domain, TRD.
-
While the genotype-phenotype correlation in females with mutations in MECP2 is hampered by X chromosome inactivation, the phenotype of males hemizygous for the same mutations should shed light on the effect of these mutations on the phenotype.
-
Our patient harbours a typical RS mutation, but unusually has survived beyond early childhood. The hypothesis that other genes are interfering with the clinical features of RS is possible.
At 2 years seizures during sleep were observed, the EEC was abnormal, and antiepileptic treatment was started, but with limited effect.
Except for an increased ratio of lactate to pyruvate in the spinal fluid all investigations, including a muscle biopsy, were normal. He has a normal male karyotype.
At 10 years old, his height is around 115 cm, weight is 24 kg, and he is microcephalic with a head circumference of 47.2 cm. He can sit unsupported, walk a few steps with good support, hold a cup, and drink with some help. He can make intense eye contact, but he seldom smiles and often has an anguished look. He rubs his eyes intensively and his eye surroundings are red and swollen. He is ataxic and spastic; he grinds his teeth, has a high pain threshold, and is often constipated. He has a severe thoracolumbar scoliosis with pronounced rotation. For the last two years he has been seizure free without treatment.
The intense eye contact raised the suspicion of RS and a mutation screen of MECP2 was performed. He fulfils all the necessary and some of the supportive criteria for RS.20
MATERIAL AND METHODS
Total genomic DNA was extracted from peripheral blood leucocytes, fibroblasts, hair roots, a skeletal muscle biopsy, and buccal cells according to standard procedures.
Mutation detection
PCR amplification of a 174 bp segment containing exon 2 and a 2430 bp segment containing exon 3, intron 3, and exon 4 of MECP2 was performed using the following primers 5′-GTTATGTCTTTAGTCTTGG-3′ (forward), 5′-TGTGTTTATCTTC AAAATGT-3′ (reverse) and 5′-CCTGCCTCTGCTCACTTGTT-3′ (forward), 5′-CGGTAAGAAAAACATCCCCAA-3′ (reverse) and Expand™ Long Template PCR System (Boehringer Mannheim).
After purification the PCR products were analysed by direct sequencing using an ABI 310 automated DNA sequencer and fluorescent dye labelled ddNTP chain terminators (Big-dye Sequencing System, PE-Biosystems) according to the manufacturer’s specifications. The sequencing primers used in this work correspond to the forward primers published by Amir et al.3
Detection of 816dup7
For direct detection of 816dup 7, PCR amplification was performed with the following primers: 5′Fam-GGCAGGAAG CGAAAAGCTGAG-3′ (forward) and 5′-CTGCACAGATCGG ATAAGAAGAC-3′ (reverse). The sizes of the normal and the 816dup7 alleles were 135 bp and 142 bp, respectively. The PCR products were analysed on an ABI-310 using the Genescan software (PE-Biosystems).
Allele specific detection of wild type MECP2 allele
We designed a specific primer 5′-AAGAAACGGGGCCG AAAGCCGGGG-3′ (forward) (bold letters indicate the sequence and position which is duplicated in the mutated allele), which in combination with a reverse primer 5′-TGAGTGGTGGTGATGGTGGTGG-3′ amplifies a 324 bp PCR product only when a normal allele is present.
To study the sensitivity of the amplification, two PCR products of 617 bp (nt 756–1372) covering the normal and the mutated allele, respectively, were cloned into two pGEM-T cloning vectors (Promega, Madison, WI) according to the manufacturer’s instructions. A serial dilution of a mixture of normal and mutant purified plasmid DNA, containing a ratio of normal to mutant DNA of 1:1 down to 1:100 000, was performed. PCR using the primer set described above was performed and the products analysed on an agarose gel.
Analysis of the MECP2 mRNA level
For RT-PCR analyses, total RNA was extracted from cultured fibroblast cell lines using RNeasy kit (Qiagen). cDNA was generated by the GeneAmp RNA-PCR kit from Perkin Elmer with MuLV reverse transcriptase and random hexamers as primers. Amplification of a 264 bp PCR product of the MECP2 cDNA was performed using intron spanning primers 5′-Hex-AGCCCGTGCAGCCATCAGCC-3′ (in exon 3) and 5′- GAGAAAGGCTTTTCCCTGGG-3′ (exon 4). Simultaneously, a 111 bp PCR product was amplified from the GAPDH cDNA using the primers 5′-Hex-TGGGGAAGGTGAAGGTCGGA-3′ (forward) and 5′-GAAGGGGTCATTGATGGCAA-3′ (reverse) spanning the splice site of GAPDH exon 1 and exon 2. The PCR products were analysed on an ABI-310 using the Genescan software (PE-Biosystems) and the ratio of the area of the peaks of MECP2 and GAPDH PCR products were calculated and used for semiquantitative determination of the expression level of the MECP2 mRNA.
RESULTS
Direct sequencing of the three coding exons and flanking intron sequences of the MECP2 identified a duplication of 7 bp (AAAGCCG) at position 816 in the transcriptional repression domain (TRD), which is located in exon 4 (fig 1). The patient’s mother did not harbour the mutation.

Sequence of the part of the MECP2 gene showing the 7 bp duplication. The duplication is underlined.
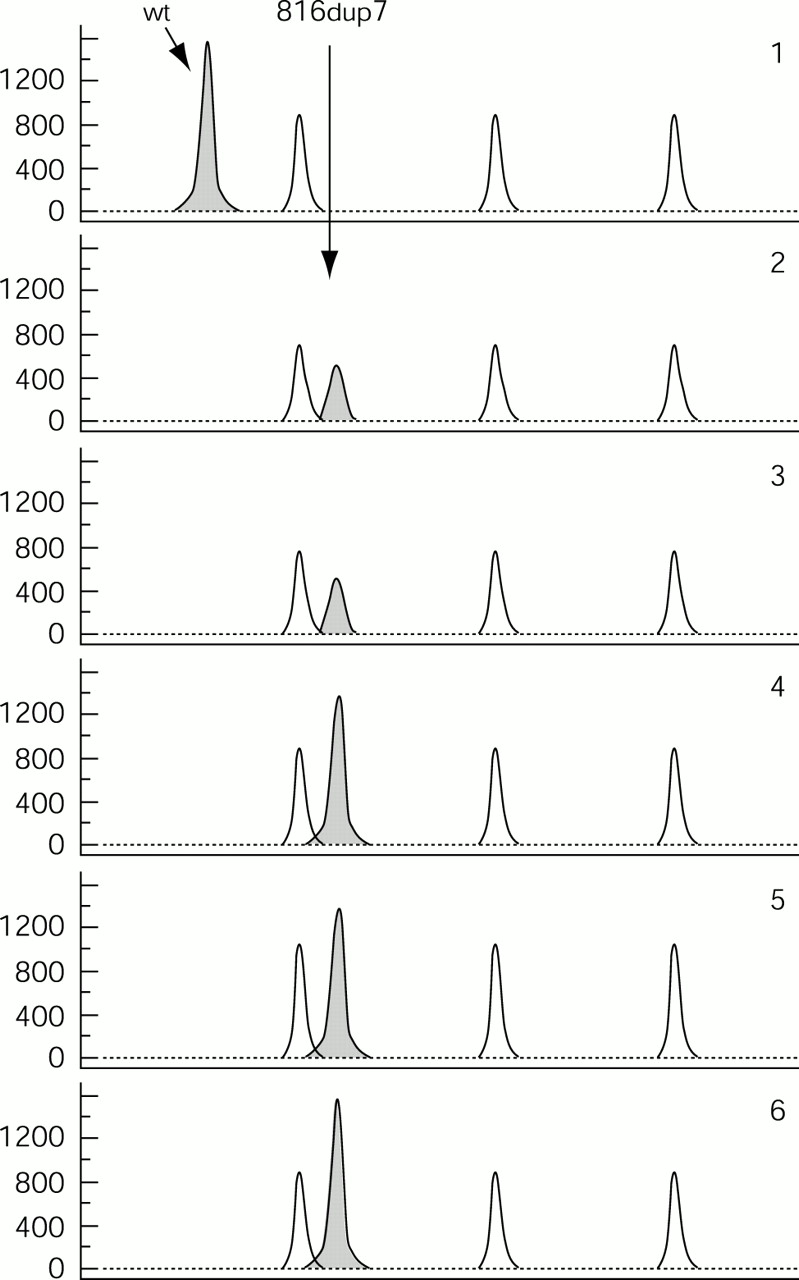
In order to test for possible mosaicism, we investigated DNA from several different tissues, including fibroblasts, hair roots, leucocytes, muscle, and buccal cells. The single appearance of the 142 bp peak in each DNA sample indicated the existence of only the mutant allele (fig 2, lanes 2 to 6).
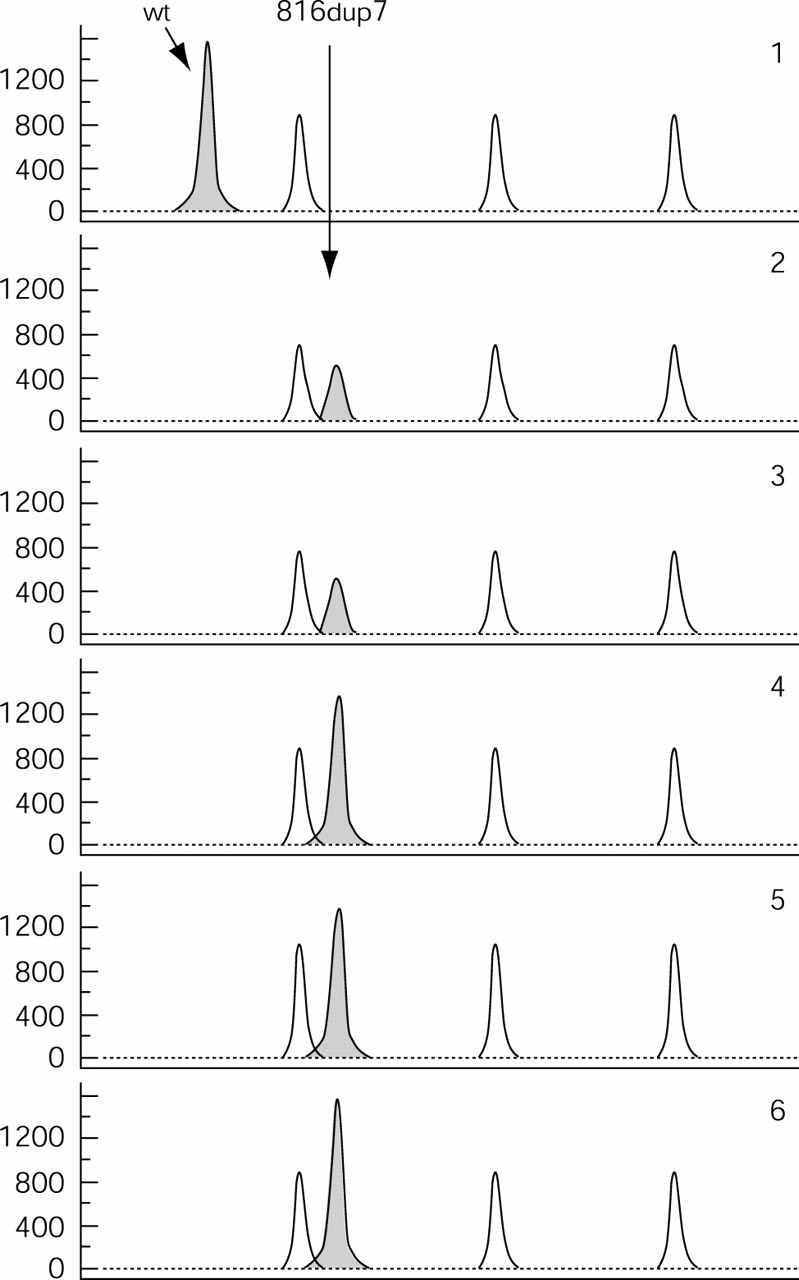
Fragment analysis for the 7 bp duplication. Arrows indicate the normal allele of 135 bases and the mutated one of 142 bases. The peaks from the GeneScan™ - 500 Rox™ size standard are also shown. Panel 1: DNA from the patient’s mother. Panels 2, 3, 4, 5, and 6: DNA from the patient’s fibroblasts, hair roots, leucocytes, muscle, and buccal cells, respectively.
To exclude any preferential amplification of the mutant allele, we designed an allele specific primer system, which shows a PCR product only if the normal allele is present. The system was able to detect fewer than 200 normal alleles among 100 000 mutant alleles (fig 3). The assay showed no sign of mosaicism in any of the tissues studied, which represent all three germ layers, the ectoderm, mesoderm, and endoderm, strongly suggesting that our patient is truly hemizygous for 816dup7.

Allele specific PCR for the normal allele. Lane 1: molecular weight marker. Lane 2: PCR amplification of 100 000 normal MECP2 alleles. Lane 3: PCR amplification of 200 normal MECP2 alleles among 100 000 mutant alleles. In this dilution, the normal allele is still detectable.
We determined the ratio between the level of MECP2 mRNA and GAPDH mRNA in cultivated fibroblasts from the patient and from five control cell lines. Our results showed that the expression of the 816dup7 allele appeared to be equal to that of the normal allele in five control fibroblast cell lines.
DISCUSSION
RS is an X linked disease almost affecting girls only, while male fetuses are believed to be aborted spontaneously. After the identification of MECP2 as the gene responsible for RS, mutations have been identified in the majority of RS patients. Mutations have been found in all parts of the gene, but some mutations seem to be recurrent. No clear correlation between genotype and phenotype has been established, although some reports7,8 seem to indicate that patients with MBD mutations (both missense and truncating) show a more severe clinical presentation than patients with missense and nonsense mutations further downstream in the gene. Mutation studies have shown MECP2 mutations in males with different clinical features, ranging from severe neonatal encephalopathy and death to mild mental retardation. The “male” MECP2 mutations reported so far can be divided into two groups (table 1): mutations which are known or strongly suspected to cause RS, when present in girls, and mutations which apparently do not affect the female phenotype or cause mild mental retardation. So far 10 patients, including our own, have been identified in the first group. These mutations are either frameshift or nonsense mutations, believed to result in the expression of a truncated protein, or the recurrent T158M and R133H mutations, which are some of the most common mutations in RS patients. Six of these nine patients fulfil all the criteria for RS. However, these patients are either mosaics for the mutation in question or have a 47,XXY karyotype and are thus not hemizygous for the mutation. Three of the nine patients died very early, because of severe neonatal encephalopathy, and thus did not have an RS phenotype. These results strongly support the idea that true RS mutations are lethal or cause very early death in hemizygous males. However, our patient is an exception as he is now 11 years old and fulfils the criteria for RS.
Male MECP2 mutations, their predicted effect on MeCP2, and the observed phenotypes in males and females
We investigated several tissues from our patient, representing the endoderm (buccal cells), ectoderm (hair roots), and the mesoderm (leucocytes, fibroblasts, and muscle), in order to detect any possible mosaicism. The assay will detect a normal allele if present down to a ratio between normal and mutant alleles of 1:200. We did not find any signs of mosaicism. We believe that this patient is a true hemizygous male, but we cannot prove that some non-mutated cells exist in the brain alone. Although the 816dup7 mutation has not been found in females, it is predicted to cause RS, since a large range of this type of frameshift mutations in the region encoding the TRD has been reported in girls with classical RS.
While predicting and comparing the genotype-phenotype correlation in females with mutation in MECP2 are hampered by X chromosome inactivation, the phenotype of males hemizygous for the same type of mutations should shed light on the pathogenic effect of these mutations. We have compared our patient’s mutation and clinical appearance with those of previously reported male patients.
One other MECP2 mutation found in a male, 806delG,9 is comparable to our 816dup7 mutation when the predicted proteins are considered (fig 4). Both mutations cause a frameshift, resulting in a nonsense codon downstream of the mutation. The corresponding mRNAs for both mutations are found in normal amounts and truncated proteins are therefore likely to be produced. The mutations are predicted to result in a truncated protein of 288 and 332 amino acids (aa), respectively. The amino acid sequence of the 816dup7 protein is correct until aa 274, and the putative nuclear localising signal (NLS) (aa 252–271) is therefore maintained, which is only partly the case for the 806delG protein, in which the correct amino acid sequence ends with aa 268. Both proteins will lack the C-terminal part and the domain believed to facilitate DNA binding. The phenotypic difference between these two patients is striking considering the similarity between the genotypes (table 1).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic presentation of the predicted MeCP2 proteins for four MECP2 mutations. The figures indicate the amino acid number. (Above) Normal MeCP2. MBD: methyl binding domain. TRD: transcriptional repression domain. C-ter: C-terminal part of the protein believed to facilitate DNA binding. NLS: Nucleic acid localisation signal. (Below) Solid lines: predicted protein with a correct aa sequence. Broken lines: incorrect aa sequence.
On the other hand, our patient shares several phenotypic signs with the patient reported by Meloni et al17 (table 2), harbouring the Q406X mutation. This patient has some RS features, but lacks some of the major criteria. He is mentally retarded and suffers from progressive spasticity. The Q406X mutation was identified in a family in which two females were heterozygous for the mutation, but presented with normal to borderline intelligence and absence of skewed X inactivation. The mutation is therefore not a classical RS mutation. The Q406X mutation is predicted to produce a protein of 405 aa, with intact TRD and NLS, lacking only part of the C-terminal region of the protein.
Comparison of signs present in female Rett syndrome, in the male with Q406X, and in the male with 816dup7 reported in the present paper
The mutation 1154del328 predicts a protein very similar to that of Q406X, but was found in a male suffering from neonatal encephalopathy, who died in early childhood.
The idea that the identification of males with mutations in MECP2 would solve the enigma of the different phenotypes associated with the mutations is not supported, and it has been suggested that other genes interfere in the development of the clinical features in these patients. MECP2 is not the only gene active in DNA methylation dependent transcriptional repression21 and variation/mutations in other genes could therefore explain the lack of genotype-phenotype correlation discussed here. Polymorphisms have been shown to modulate the phenotypic expression of disease mutations in other genes, for example, CFTR,22,23 and very little is known about variation in the introns in MECP2 or the importance of the alternative MECP2 transcripts.
The patient presented here adds to the variation in the phenotypic expression of mutations in MECP2, ranging from severe encephalopathy and neonatal death in classical RS to mild mental retardation found in both females and males.
Acknowledgments
This work was supported by grants from the Novo Nordic Foundation, The Gangsted-Rasmussen Foundation, Dagmar Marshall Foundation, and Signe and Peter Gregersen Foundation.