Article Text

Statistics from Altmetric.com
Editor—Canavan disease (McKusick No 271900) is an autosomal recessive disorder characterised by spongy degeneration of the brain. This disorder is most prevalent in people of Ashkenazi Jewish descent but has been observed in other ethnic groups. Patients have severe mental retardation, poor head control, macrocephaly, and seizures. Canavan disease is caused by the accumulation of N-acetylaspartic acid (NAA) in the brain as the result of a deficiency of aspartoacylase (ASPA) (EC 3.5.1.15) activity.1
The human ASPA gene has been mapped to chromosome 17p13. The gene spans a distance of approximately 30 kb and is organised into six exons.2 Previously described mutations in the ASPA gene include single nucleotide changes leading to missense, nonsense, and exon skipping mutations and frameshift mutations caused by the insertion or deletion of a small number (1-4 bp) of nucleotides.3 In Ashkenazi Jewish patients, the E285, Y231X, and 433-2(A to G) mutations account for approximately 82.9%, 14.8%, and 1.1% of disease alleles respectively. The predominant mutation in non-Jewish patients is the A305E mutation, which accounts for approximately 60% of non-Jewish Canavan disease mutations.4 Surprisingly, large deletions involving the ASPA gene have not yet been described. Here, we report the first instance of a partial deletion of the ASPA gene in Canavan disease. Two affected children in a family from Mexico were homozygous for a 190 kb deletion. The parents of the affected sibs were second cousins once removed. There was no other family history of Canavan disease or other neurodegenerative disorder.
Testing of the affected children for the common Jewish and non-Jewish Canavan disease mutations was negative. Preliminary Southern blot experiments using an ASPA cDNA suggested that the affected children were homozygous for a deletion that included at least the first two exons of the ASPAgene (data not shown). To confirm this result, the first three introns of the ASPA gene were amplified from genomic DNA. Amplifications were performed using the GeneAmp XL PCR kit (PE Applied Biosystems) and primers located in the flanking exons (intron 1: EX1F 5′-GCATTGGCTAGAGAATGGCGCT-3′ and EX2R 5′-CCCATGTTAGAGGTGGTGTTGTGAAG-3′; intron 2: EX2F 5′-CCTTCACAACACCACCTCTAACATGG-3′ and EX3R 5′-GCTATGGAACGAGTGGTCGCAT-3′ intron 3: EX3F 5′-CGACCACTCGTTCCATAGCCA-3′ and EX4R 5′-CAGAACCCCTTGAGGCTGAGGA-3′). All three introns were successfully amplified using DNA from the parents and a normal control. However, only intron 3 was amplified when DNA from the affected children was used (data not shown), suggesting that the affected children were homozygous for a partial deletion of theASPA gene. This result was confirmed by a duplex PCR reaction in which introns 2 and 3 were simultaneously amplified. As expected, both introns were detected in the parents but only intron 3 was present in the affected children (data not shown).
Southern blot analysis was then carried out to identify a junction fragment spanning the deletion breakpoints. Since the proximal breakpoint of the deletion was likely to be in intron 2, a 500 bpStuI fragment from the 3′ end of intron 2 was used as a probe. With BamHI digested DNA from a normal control, the probe hybridised to a 19.8 kb fragment (fig1). This 19.8 kb fragment was replaced by a 2.1 kb fragment in the affected children. In the parents, both the 19.8 kb and 2.1 kbBamHI fragments were detected. A similar pattern of normal and abnormal restriction fragments was observed withHindIII (fig 1) andEcoRI (data not shown). The fact that the abnormal hybridising fragments were observed with multiple restriction enzymes indicated that these fragments represented deletion junction fragments and not restriction fragment length polymorphisms.
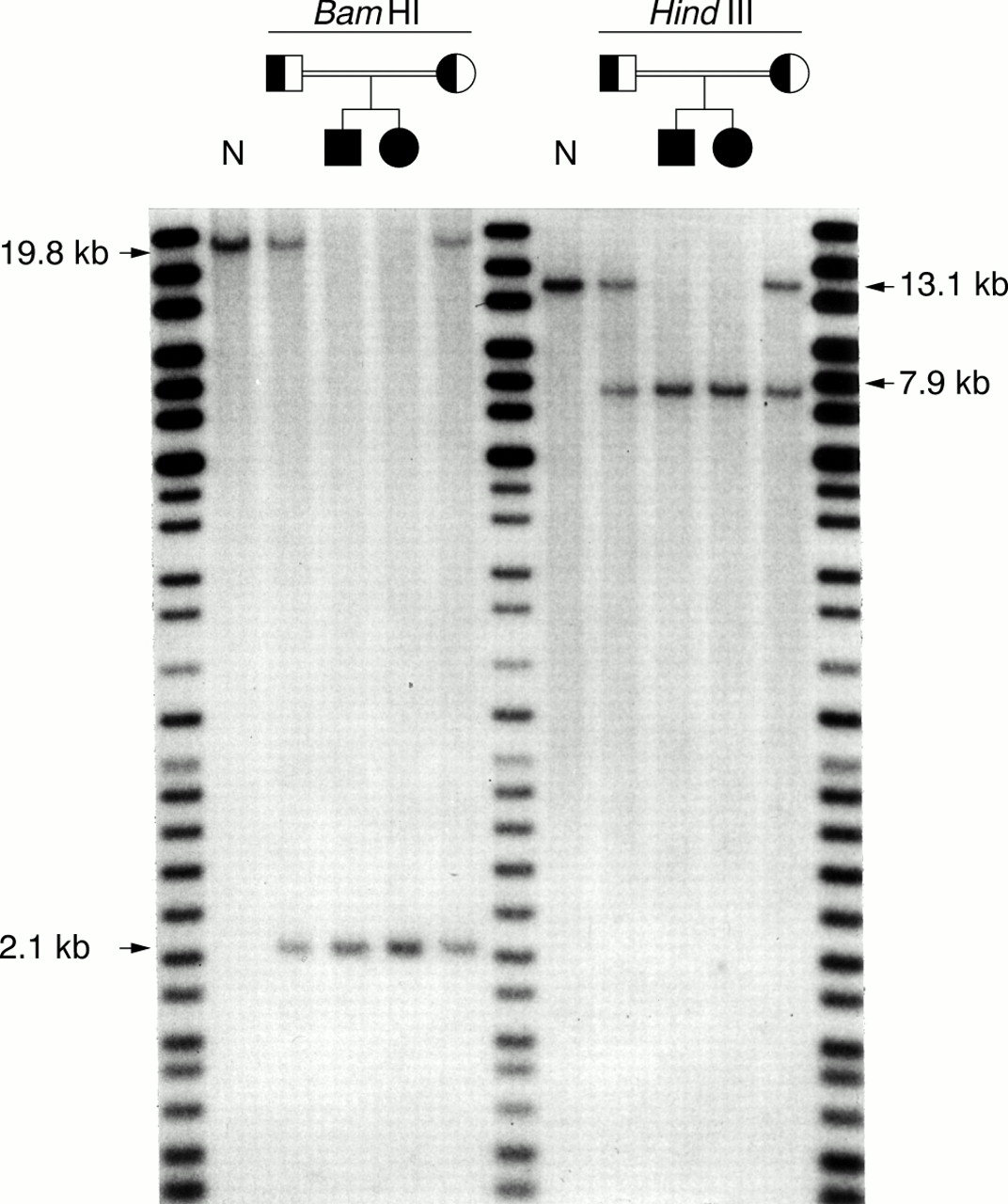
Identification of junction fragments by Southern blot analysis. Genomic DNA was digested with either BamHI or HindIII and hybridised with a 500 bp StuI fragment from the 3′ end of ASPA intron 2. Both affected children were homozygous for either a 2.1 kb BamHI or 7.9 kb HindIII junction fragment. The normal 19.8 kb BamHI and 13.1 kb HindIII fragments were not observed. The parents carried both the normal and abnormal hybridising fragments. N, normal control. The DNA Analysis Marker System (Gibco BRL) was run as a molecular size standard.
To identify precisely the location of the proximal and distal deletion breakpoints, the inverse PCR technique5 was used to isolate the 2.1 kb BamHI junction fragment for nucleotide sequence analysis. For inverse PCR of the junction fragment, genomic DNA (2.5 μg) was digested withBamHI and restriction fragments ranging in size from 1.5 kb to 3.0 kb were gel purified. The purified DNA was then circularised in 20 μl reactions containing 100 ng of DNA and 0.8 units of T4 DNA ligase. Inverse PCR amplifications were performed using outwardly oriented primers derived from exon 3 (EX3F and EX3inv 5′- ACGTAGCAGGGTAGTGGAGCCAG-3′) and the GeneAmp XL PCR kit. Since the region between the primers was not amplified, a 1.9 kb product was expected. As expected, inverse PCR amplification ofBamHI digested DNA from an affected child resulted in the synthesis of a 1.9 kb product. No amplification product was observed using DNA from a normal control (data not shown).
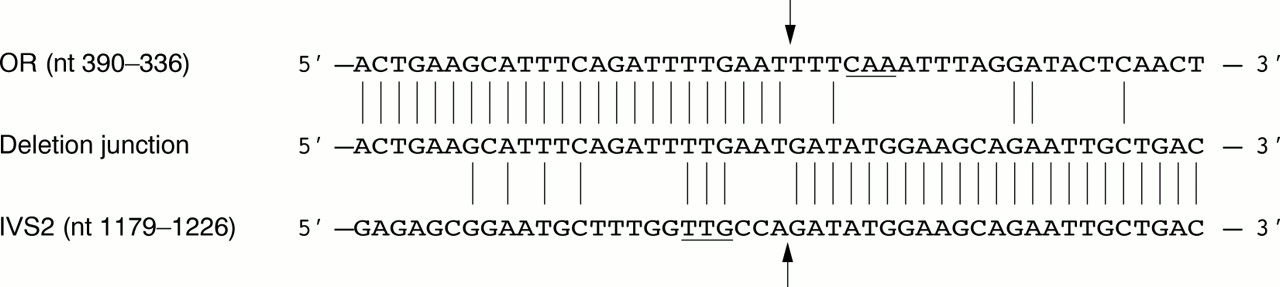
The 1.9 kb inverse PCR product was sequenced in its entirety. A comparison of this sequence with that of introns 2 (GenBank Accession No AF276424) and 3 (data not shown) showed that the proximal deletion breakpoint was in intron 2, 499 bp upstream of exon 3. A search of the GenBank nucleotide sequence database indicated that the distal deletion breakpoint was located approximately 19.9 kb downstream of the OR17-93 (OR1E2) gene, the most centromeric member of the chromosome 17p13.3 olfactory receptor (OR) gene cluster.6 Since the distal deletion breakpoint was outside the OR gene cluster, the deletion did not include any of the OR coding regions. The sequences of the proximal and distal deletion breakpoints are shown in fig 2.

{kind=link}
{kind=link}
Nucleotide sequence of the deletion breakpoints. The sequences of the olfactory receptor gene cluster (OR), the deletion junction, and intron 2 of the ASPA gene (IVS2) are aligned. Sequences are numbered according to GenBank Accession Nos AC007194 (OR) andAF276424 (IVS2). Vertical lines indicate positions at which the sequences are identical. The locations of the distal and proximal deletion breakpoints are indicated by arrows. A 3 bp inverted repeat located 3 bp from the deletion breakpoints is underlined.
An estimate of the size of the deletion was made on the basis of a previously published physical map of the 17p13 region.7The ASPA gene and the OR17-4 (OR1D2) gene (previously designated theOLFR1 gene) were mapped close to the centromeric and telomeric ends, respectively, of the CEPH YAC clone 898A10. As the size of this YAC clone is approximately 570 kb, we estimated that this was the maximum distance between theASPA and OR17-4 genes. The nucleotide sequence of the OR gene cluster (GenBank Accession No AC007194) indicated that the OR17-4 gene is located approximately 377 kb upstream of the distal deletion breakpoint. Since the distance between theASPA and OR17-4 genes was estimated to be approximately 570 kb, we estimated that the deletion had a maximum size of approximately 190 kb.
The fact that the affected children have a clinical presentation that is typical for Canavan disease indicates that the 190 kb interval between the ASPA gene and the telomeric OR gene cluster probably does not include any essential or non-redundant genes. Any genes in this interval would be homozygously deleted in the affected children and the absence of these genes would probably be reflected in the phenotype of the children.
The mechanism by which the deletion arose in this family is unknown. No sequence homology between the ASPA gene and the OR gene cluster in the region of the deletion breakpoints was found, except for a 3 bp inverted repeat (CAA/TTG) 3 bp from both the distal and proximal breakpoints (fig 2). The deletion therefore appears to have been the result of a non-homologous recombination event.
We have shown that patients with Canavan disease may carry large deletions involving the aspartoacylase gene. The spectrum of mutations responsible for Canavan disease must therefore be expanded to include this type of mutation. The prevalence of deletion mutations in Canavan disease is at present unknown but is likely to be low, as the majority of Canavan disease patients carry point mutations in theASPA gene. However, our results indicate that deletions of the ASPA gene should be considered as a cause of Canavan disease whenASPA point mutations cannot be identified.