Article Text

Statistics from Altmetric.com
Editor—Inherited mutations in theE-cadherin gene (CDH1) were first described in three Maori kindreds with early onset, diffuse, familial gastric cancer.1 More recently, this finding has been confirmed in other populations2-7 and this dominantly inherited familial cancer syndrome has been designated a hereditary diffuse gastric cancer (HDGC).4 So far, no germline mutations have been identified in site specific intestinal type gastric cancer. Based on the guidelines of the First Workshop of the International Gastric Cancer Linkage Consortium (IGCLC), the following criteria were introduced: (1) two or more documented cases of diffuse gastric cancer in first/second degree relatives, with at least one diagnosed before the age of 50 or (2) three or more cases of documented diffuse gastric cancer in first/second degree relatives, independently of age of onset. In addition, criteria for familial intestinal gastric cancer (FIGC) were defined.8
In the present study, we analysed 11 Finnish gastric cancer patients with a family history of disease and two sporadic cases with germlineE-cadherin gene mutations (table 1, fig 1). None of these families completely fulfilled the criteria for other inherited cancer syndromes with predisposition to gastric cancer, for example, hereditary non-polyposis colorectal syndrome (HNPCC), familial adenomatous polyposis (FAP), Peutz-Jeghers syndrome, or Li-Fraumeni syndrome (LFS).8 ,9 Five of the families studied fulfilled the criteria for HDGC syndrome (table 1, fig 1, Nos 1-3, 5, and 10) and five families included two or more cases of gastric cancer (one of which was confirmed to be of diffuse type) (table 1, fig1, Nos 4, 6, 9, 11, and 12). Family 13 included four intestinal type gastric cancer cases and therefore seems to belong to FIGC. However, one of the patients in this family had diffuse type carcinoma. In addition to gastric cancer, 11 families also displayed other cancer types.
Features of the families studied

Pedigrees of gastric cancer families. Patients analysed in this study are marked by an asterisk. An arrow depicts the person carrying the P172R change (family 1). Bas, basalioma; Bla, bladder cancer; Br, breast cancer; CRC, colorectal cancer; Ga, gastric cancer; Kid, kidney cancer; Leu, leukaemia; Lip, lip cancer; Liv, liver cancer; Lu, lung cancer; Mel, melanoma; Ov, ovarian cancer; Pan, pancreas cancer; Pro, prostate cancer; Sar, sarcoma; Ski, skin cancer; Thy, thyroid cancer; Un, unknown; Ut, uterine cancer. The age at diagnosis, when known, is shown in parentheses.
E-cadherin mutation analysis was performed by genomic sequencing of the 16 coding exons including exon/intron boundaries. DNA from one patient with gastric cancer from each of the families was isolated according to standard procedures. Exons were amplified using primers described by Berx et al,10 except exons 4 and 5, which were amplified as described in Gayther et al.2 The reactions were carried out in a 50 μl reaction volume containing 100 ng of genomic DNA, PCR buffer (PE/ABI, Foster City, CA), 200 μmol/l each dNTP (Finnzymes, Espoo, Finland), 0.6 μmol/l each primer, and 1 unit AmpliTaq GOLD polymerase (PE/ABI). The concentrations of MgCl2 in the reaction mixture were as described by Berx et al,10except that for exon 6 the concentration of MgCl2 was 1.5 mmol/l and for exon 1 DMSO (5%) was included in the reaction mixture. PCR reactions were carried out as described in Berxet al 10 with the following changes: annealing temperature for exon 2 was 54°C, for exon 6 56°C, for exon 8 54°C, and for exon 13 57°C. Sequencing reactions containing 40 ng of the PCR product with 3.2 pmol of the sequencing primer in a volume of 12 μl were performed using ABI Prism Dye Terminator Cycle Sequencing Ready Reaction Kit (Perkin-Elmer, Foster City, CA) or ABI Big Dye Terminator Kit (Perkin-Elmer, Foster City, CA) according to the manufacturer's instructions. Sequencing reactions were electrophoresed either on 6% Long Ranger gels, containing 8 mol/l urea, or 5% Long Ranger gels, containing 6 mol/l urea, and analysed on an Applied Biosystems model 373A or 377 automated DNA sequencers, respectively.
Exons 1 and 4 were amplified as described above and analysed using SSCP from 84 and 212 cancer free controls, respectively. After PCR, 5 μl of each sample was mixed with 5 μl of denaturing loading buffer (95% formamide, 20 mmol/l EDTA, 0.05% bromphenol blue, 0.05% xylene cyanole FF), denaturated for five minutes at 94°C and loaded into a 0.4 mm × 30 cm × 45 cm gel. Electrophoresis was performed for exon 1 using gels containing 0.5 × MDE solution (AT Biochem, Malvern, PA) and 0.6 × TBE buffer and were run at 4 W for 20 hours. Exon 4 was analysed using 1 × MDE solution and 2.5 mol/l urea at 6 W for 14.5 hours. The gels were silver stained according to standard procedures.
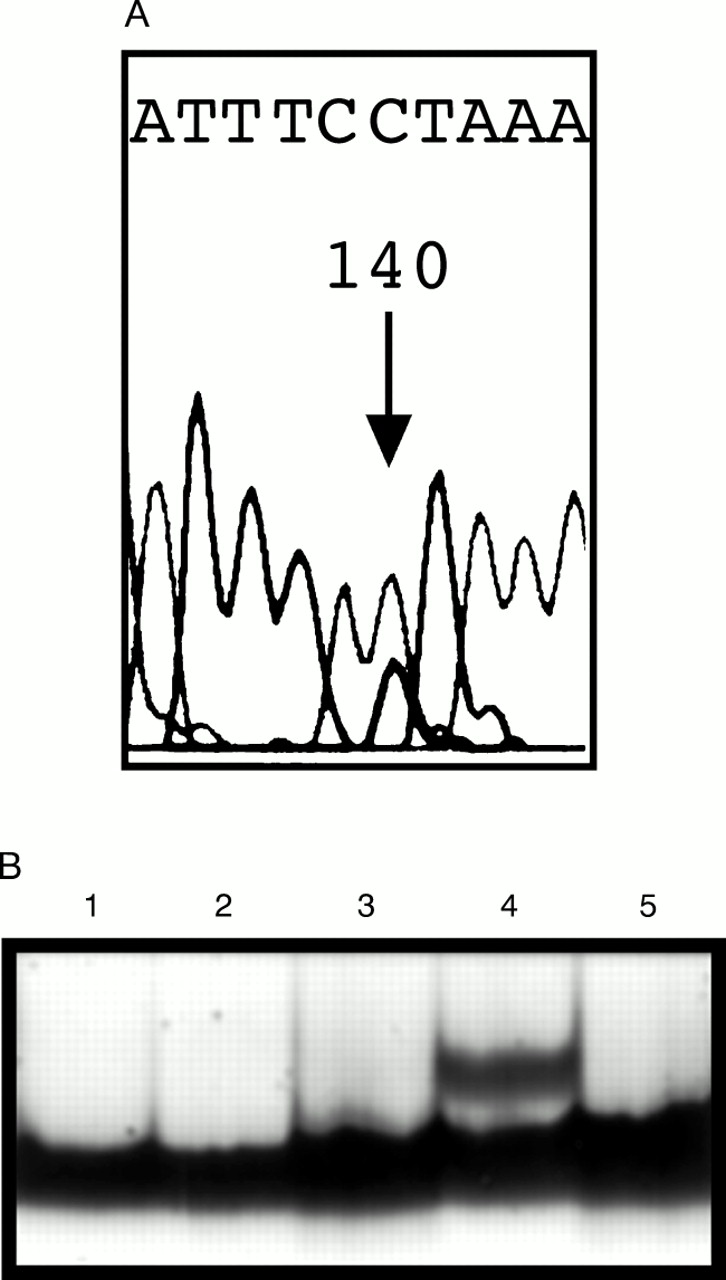
We detected one potential missense mutation in the codingE-cadherin gene sequence (table 1, fig 1, No 1). A C to G change occurred in codon 172 in exon 4 resulting in substitution of proline by arginine (P172R) (fig 2A). This family contains seven gastric cancer cases in three different generations. Three of the affected subjects had gastric cancer under 50 years of age (33, 39, and 40 years). One of them also had ductal breast cancer. In addition, one patient with both bladder and ovarian cancer and another with prostate cancer and basalioma were found in this family. To investigate the segregation of this missense type change in the family, we screened two additional family members with gastric cancer (fig 1). DNA from paraffin embedded tissues was isolated according to standard procedures and mutation analysis was performed as described above. However, neither of them carried the P172R change. One of the patients studied was the mother of the mutation carrier. The father of this patient died at the age of 94 years and was cancer free. This change was also absent in 212 control samples from cancer free subjects, as screened by SSCP analysis (fig 2B). The change appears to be a rare polymorphism.

{kind=link}
{kind=link}
(A) Direct sequencing shows a heterozygous C→G change (P172R, see arrow). (B) SSCP analysis of the P172R change. A positive control (lane 4) was included in all SSCP runs.
Four additional polymorphisms of theE-cadherin gene were found in this series of gastric cancer patients. A C to T silent change in codon 692 (from alanine to alanine) occurred in eight of 13 (61.5%) gastric cancer patients. A C to T change in codon 751, resulting in aspartate substitution by asparagine, was detected in three of 13 (23%) patients. These two polymorphisms have been previously reported.10 ,11 A C to G change was found before the start codon (−71 bp) in the non-coding region in one of 13 (7.7%) gastric cancer patients and in two of 51 (3.9%) cancer free controls. A T to C change at position +6 in intron 1 occurred in five of 13 (38%) gastric cancer patients and in 18 of 51 (35%) cancer free controls.
So far, altogether 14 truncatingE-cadherin germline mutations have been detected in gastric cancer patients.8 A few putative missense germline mutations have been reported but their functional significance has not been tested.1 ,6 ,7 In the sporadic type of cancer there seems to be a cluster of mutations between exons 7 and 9 whereas germline mutations are more evenly distributed.8 ,11 A novel missense type change, P172R, found in this study is located in exon 4 which encodes a large extracellular domain with Ca2+ binding motifs (exons 4-13).10 Based on the segregation of the mutation in affected cases in this particular family, it seems that this change is not a pathogenic mutation. It seems to be a very rare polymorphism because none of the 212 cancer free controls carries this change. This finding is interesting because altogether seven gastric cancer cases were found in this family. Caldas et al 8 have suggested thatE-cadherin should account for 25% of the families fulfilling the established criteria for HDGC. However, PCR based screening methods used in this study do not allow detection of all mutation types, for example, large deletions.
Our results support the notion that germline mutations in theE-cadherin gene are responsible for only a subset of gastric cancer patients with a family history of the disease. In our study, no mutations were found in 13 gastric cancer probands. Five of the families studied fulfil the criteria for HDGC and one for FIGC. Our data suggest that for the purpose of efficientE-cadherin mutation detection, there may be a need for more stringent criteria for HDGC, such as requiring three affected subjects as is common in research on familial breast and colon cancer. However, our data set is limited. Loose inclusion criteria should encourage collection of gastric cancer families. This is important, because further work is necessary to identify predisposing gene(s) for a subset of HDGC families, as well as families segregating intestinal gastric cancer.
Acknowledgments
We thank Inga-Lill Svedberg, Tuula Lehtinen, Sinikka Lindh, Annika Lahti, and Kirsi Laukkanen for technical assistance.