Article Text

Statistics from Altmetric.com
Somatic mosaicism is the increasingly recognised phenomenon where there are two populations of cells that differ genetically in the same individual. When one considers that there are approximately 1014 cells in the adult human body, about 30 000 genes and approximately 10−6–10−7 mutations per gene per cell division, it follows that all of us are mosaic at some level, for some genes. Recently, two reports, by Hes et al1 in this issue (see page 71) and by Aretz et al,2 demonstrated that somatic mosaicism can be found in about 20% of sporadic cases of familial adenomatous polyposis coli (FAP) in which mutation of APC can be found. Genetic testing of the affected individuals revealed mutation, but at a level that accounted for less than all copies of the implicated allele, suggesting that some cells and/or tissues had the mutation while others did not.
Mosaicism of NF1 and NF2 has been reported several times.3 Mutations are only found in pathologically affected tissues, and the clinical features and course of disease are often milder than usual.4 5 This is presumably because not all the susceptible tissues have inherited the causative mutation. Mosaicism has also be reported in Marfan syndrome, tuberous sclerosis, achrondroplasia, Duchenne muscular dystrophy and retinoblastoma among many other conditions.6 7
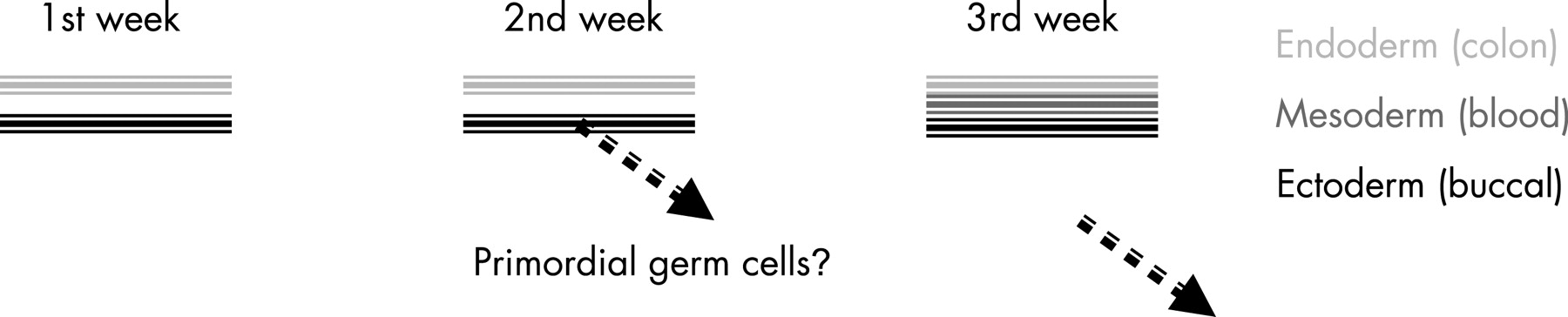
The point in embryological development at which a mutation arises has profound consequences for the individual and the family. If it arises in an essential domain of the APC gene in a single colonic epithelial stem cell, the only consequence will probably be adenomatous polyps in the segment of the colon that eventually becomes populated with the descendants of that single stem cell (eg, as hypothesised in the discussion of patient 7 in the series presented by Hes et al1). Surgery may then be required, but future descendants will not inherit the mutation (see fig 1A). If the mutation arises earlier, in an endoderm cell that pre-dates the commitment to cell fate of the six or so stem cells that will populate the adult colonic epithelium, then the entire colon will be affected. In such a case, the consequence will be classic FAP but without extracolonic features such as osteomas, dentition anomalies and desmoids. Should the mutation arise earlier again, before gastrulation, or the formation of the epiblast (which pre-dates the formation of the endoderm and mesoderm layers), it may be included, not only in the future colonic epithelium, but possibly also in the ectodermal layer. This would have at least two consequences: the first would be that all of the mesoderm would be affected, and mutations would be detectable, at some level, in peripheral blood cells arising from the mesoderm. The second would be that as the primordial germ cells (the future germline of the individual) migrate from what we believe is the ectodermal layer (based on our knowledge of mouse embryology during the second week of development), these early mutations may also be found in reproductive cells and, if so, would therefore be transmissible to the next generation in the family.

Depending on the level and distribution of mosaicism, the degree of severity of the condition may also be greatly underestimated for offspring; later mutations that are still early enough to be included in the future germline (reproductive cells) may only affect some, but not all of the future colonic epithelial stem cells (see fig 1B). Then the clinical results may be a milder, attenuated form of FAP, due to the competing population of normal and mutated colonic epithelial stem cells, or segmental FAP, possibly affecting only a discrete portion of the colon. However, mutations that are then transmitted via the germline will affect all of the cells of the individual in the next generation, causing their clinical presentation to be more similar to that generally seen with FAP mutations.
If the mutation arises only in the primordial germ cells, after they have separated from the developing germinal discs, the consequences are unique. The clinical features of the disease are not manifested in the developing individual, even as they age, because their somatic tissues (including colonic cells) have not developed the mutation. However, their germline will be affected and they will be predisposed to pass the mutation on to their children with a probability depending on the stage at which the mutation developed (see fig 1C). Their affected children will have a constitutional mutation, and will appear to be the first affected patient, the index case, in the family, with an apparently de novo mutation. These situations are unremarkable unless or until a sibling is found to have the same mutation, which neither of the parents share (by standard testing). For this reason, it is believed that germline mosaicism is under-recognised. However, in these rare cases, the risks for other siblings also being unknowingly affected with the same condition would also be greatly underestimated.</emph>8
As analysis of DNA from circulating lymphocytes isolated from peripheral blood is the standard mutation detection method, mutations arising at later stages in an individual’s embryonic development will not be detectible by routine testing and analysis. This is where the results of the paper by Hes et al1 help the clinical picture. They extract from the pool of what has until now been classified as “APC-normal; no mutation found”, “an intermediate”; a mosaic; a person with a mixture of two different genetic populations of cells. Their data are also consistent with a recently published comprehensive analysis of 1248 FAP patients from the German Registry, in which six out of eight cases of somatic mosaicism were demonstrated to be associated with an attenuated phenotype.2
The paper by Hes et al1 addresses these fundamental questions and highlights some of the consequences. Within the data set examined by the authors, approximately 20% of apparently sporadic cases of FAP were shown to display mosaicism on sequence analysis of peripheral blood samples, and/or adenomas. This is an enormous improvement on the current detection rates of mutations in clinically diagnosed patients, particularly the apparently sporadic and the proven de novo groups. Having answers for these cases is not only intellectually satisfying, but it obviates the clinical necessity for further testing. This issue raises questions about the increased level of data analysis that would be necessary to identify these patients, and differentiate them from routine cases. Different commercial testing laboratories have differing proprietary software analysis packages that are not currently optimised to recognise these cases from background noise, without manual readers.
What Hes et al do not directly address, but is of interest, is where do the remaining 80% of identified mutations in sporadic cases of FAP arise? The 20% represent those caught in the window between when they first arose and when they become either “fixed”—that is, established as transmissible mutations—or “lost”—that is, not transmitted to subsequent generations. The remaining 80% must then be a combination of those inherited from the previous generation as de novo mutations arising in the parental germline (as case 6 of table 1 of Hes et al1) and those arising so early in the embryological development of the sequenced individual as to account for close to all of the peripheral blood stem cells as well as the colonic epithelium. Put another way, given that every new mutation must arise in a single individual, these must either arise as very early somatic mutations such that the tested individual appears to have a constitutional mutation or they are germline mutations from the previous generation (see fig 1).
The timing and tissue of origin of the de novo mutation during embryogenesis will have consequences for whether and with what frequency the mutation may be transmitted to subsequent generations. As the endoderm and ectoderm are distinct by day 8 of development, and the mesoderm develops from the primitive streak by the third week, somatic mutations have a narrow window of opportunity within which they must arise to be included in the tissue that is fated to become the germinal (reproductive) layer. As the primordial germ cells are presumed to separate from the ectodermal layer of the embryo during the second week of development,9 10 it follows that some early mutations are destined to become established in the germline of the developing embryo, while other later mutations, not fated for inclusion in the mesoderm or ectoderm, may not be either detectable by genetic testing of peripheral blood or transmissible to the next generation, or both.11 See fig 2 for a simplified summary of the timeline of germinal layer formation and primordial germ cell generation. These cases may account for at least some of the approximately 49% (295 out of 599) of FAP patients reported by Hes et al1 and others12 13 who fail to reveal even subtle signs of mutation, in either APC or MYH. Contrary to the prevailing standard of practice, there may be a strong rationale for re-ordering genetic testing on affected offspring of the index case in a mutation-negative family in the case of an unidentified mutation.

{kind=link}
{kind=link}
In some conditions, a causative mutation can progress, resulting in an earlier age of onset and increased severity of manifestations in successive generations, by what is known as anticipation. This phenomenon is not generally noted as a feature of FAP/AFAP. There have been occasional reports suggesting documented cases,14 15 as well as studies attributing such observations to artefactual causes.16 Anticipation has been observed in association with mosaicism in several conditions, including osteogenesis imperfecta, Ehlers–Danlos syndrome, congenital contractural arachnodactyly and Marfan syndrome17 18 The most telling specific question, whether de novo attenuated cases of FAP, when transmitted, tend to give rise to cases of classical FAP, remains to be addressed. As Hes et al1 point out, the most efficient way of detecting these families will be first to offer genetic testing to affected grandchildren in multigenerational families—that is, to the second clinically affected generation rather than the first, where possible. This strategy will circumvent false negatives due to mutations not present in the blood of the first clinically affected generation in the family (see fig 1B)
The other intriguing aspect of mosaicism and the origin of mutations that Hes et al1 address is the propensity for certain types of mutation to arise under certain circumstances; they observe a statistically significant excess of C>T transitions among their somatic mosaic mutations (an observation independently made in the German Registry). Interestingly, differences in the mutation profile of somatic versus germline mutations have been reported for NF119 and NF2.20 In a similar vein, de novo APC point mutations and large (but not small) deletions have been reported to arise exclusively on the paternal chromosome, albeit in a study using small numbers,21 an observation mostly in agreement with previous findings using a different patient pool.22 Further analysis of somatic versus germline-derived mosaic mutations may help to segregate these mutations into separate classes.
These observations open the question of how mutation and/or stringency of error-proofing pathways may be physiologically regulated in mitosis and meiosis. Further analysis of these types of observations may help build a Bayesian model that incorporates individuals’ specific molecular, medical and family histories into a risk assessment model for family members of probands in whom apparent somatic mutations are detected. As they carve out future directions for improved diagnostics, Hes et al are poised to incorporate several of these anomalies into their detection algorithm.
For clinicians, a previously esoteric subject now has several important practical applications:
Polyposis patients, especially those with affected children whose genetic test results are negative, may not be truly negative, but may be mosaic.
The old adage of “if we can’t find a mutation in you, we won’t find it in your children” may not always be true: it is justified to test the second generation if there are affected individuals. For this reason, testing a younger affected generation, if available, is preferable to testing the first affected generation, or index case.
When a mosaic mutation is identified in a case of attenuated FAP, any affected children may be more severely affected, due to having the mutation in all, rather than in just some of their cells and tissues.
We do not yet know what proportion of sporadic cases of FAP, that are negative for a genetic mutation, are heritable. Therefore, counselling for reproductive risk is difficult, but must include the possibility of up to 50% risk for each child.
Hes et al have highlighted these issues, not only for FAP, but for dominantly inherited conditions in general. We look forward to the next chapter in this developing story.
REFERENCES
Footnotes
Competing interests: None.