Article Text

Abstract
Background Fanconi anaemia (FA) is a heterogeneous inherited disorder clinically characterised by progressive bone marrow failure, congenital anomalies and a predisposition to malignancies.
Objective Determine, based on correction of cellular phenotypes, whether XRCC2 is a FA gene.
Methods Cells (900677A) from a previously identified patient with biallelic mutation of XRCC2, among other mutations, were genetically complemented with wild-type XRCC2.
Results Wild-type XRCC2 corrects each of three phenotypes characteristic of FA cells, all related to the repair of DNA interstrand crosslinks, including increased sensitivity to mitomycin C (MMC), chromosome breakage and G2–M accumulation in the cell cycle. Further, the p.R215X mutant of XRCC2, which is harboured by the patient, is unstable. This provides an explanation for the pathogenesis of this mutant, as does the fact that 900677A cells have reduced levels of other proteins in the XRCC2–RAD51B-C-D complex. Also, FANCD2 monoubiquitination and foci formation, but not assembly of RAD51 foci, are normal in 900677A cells. Thus, XRCC2 acts late in the FA–BRCA pathway as also suggested by hypersensitivity of 900677A cells to ionising radiation. These cells also share milder sensitivities towards olaparib and formaldehyde with certain other FA cells.
Conclusions XRCC2/FANCU is a FA gene, as is another RAD51 paralog gene, RAD51C/FANCO. Notably, similar to a subset of FA genes that act downstream of FANCD2, biallelic mutation of XRCC2/FANCU has not been associated with bone marrow failure. Taken together, our results yield important insights into phenotypes related to FA and its genetic origins.
- Fanconi anemia
- XRCC2
- RAD51 paralogs
- DNA interstrand crosslinks
- Breast cancer susceptibility
Statistics from Altmetric.com
Introduction
Fanconi anaemia (FA) is the most frequent inherited bone marrow failure (BMF) syndrome and is clinically characterised by progressive BMF, congenital anomalies and a predisposition to acute myeloid leukaemia and other malignancies.1 ,2 The failure of the haematopoietic system on average manifests around 8 years of age, but this is highly variable, among different FA subgroups and also within the same subgroup, partly depending on the severity of the mutation.3 ,4 In addition, as many as 30% of patients with FA do not show any congenital abnormalities, which can include radial ray abnormalities, short stature, microcephaly, abnormal pigmentation and hypogonadism.3 This further demonstrates that, phenotypically, FA is a pleiotropic disease.
Additionally, FA is typified by exquisite hypersensitivity of cells to DNA interstrand crosslinking (ICL) agents such as diepoxybutane (DEB) or mitomycin C (MMC).2 ,5 ,6 Cells from patients with FA also characteristically display other phenotypes that are associated with a defective response to DNA interstrand crosslinkers, including chromosomal abnormalities and cell-cycle defects.5 ,6 These cellular phenotypes extend across all FA genetic complementation groups.1
At present, germ-line defects in 19 mostly recessive genes are clinically associated with FA. FANCR/RAD51, FANCS/BRCA1 and FANCT/UBE2T are the most recently identified FA genes.7–12 The products of these genes interact in different protein complexes that comprise the FA-breast cancer susceptibility (BRCA) pathway, which together, are essential for the error-free removal of DNA ICLs.2 ,13
The products of eight of the FA genes (FANCA/-B/-C/-E/-F/-G/-L/-M) form the FA core complex. This large protein complex functions as an E3 ligase and, in combination with the FANCT/UBE2T E2 conjugase, culminates in the monoubiquitination and activation of FANCD2 and FANCI,7–9 ,14–16 a central step in the FA pathway. Loss-of-function defects in any of these so-called ‘early’ or ‘upstream’ genes leads to a defect in the monoubiquitination of FANCD2 and FANCI.6 ,14–16
The other eight FA genes are classified as ‘late’ or ‘downstream’ FA genes because cells that are deficient in these genes have intact FANCD2 monoubiquitination.13 A subset of FA proteins, all of which are products of late FA genes, including BRCA1/FANCS, BRCA2/FANCD1, PALB2/FANCN and RAD51C/FANCO, have a common role in regulating the assembly of RAD51 foci.10 ,17–19 Further, some of these late FA genes, including FANCO/RAD51C, FANCR/RAD51 and FANCS/BRCA1, are atypical, as patients with germline mutations in these genes show FA-related malformations but do not seem to develop BMF.8 ,10 ,18
We previously reported a child with congenital abnormalities suggestive of FA. At the age of 2.5 years, the boy displayed normal bone marrow cellularity and normal peripheral blood values.20 Based on positional mapping and whole exome sequencing, the child was found to have inherited, among other germ-line mutations, a premature biallelic stop gain mutation in XRCC2.20 While these phenotypes suggest that XRCC2 could be an FA gene, this hypothesis remains to be critically tested.
Here, on the basis of complementation of three different FA-related cellular phenotypes stemming from defective repair of ICLs, we demonstrate that XRCC2 is the 20th FA gene and is the second RAD51 paralog so identified. We also provide insight into the function of XRCC2 in preventing FA by demonstrating that it is required to maintain levels of another FA protein, RAD51C, as well as other RAD51 paralogs that are normally present in the XRCC2-RAD51B-C-D complex. Taken together, including the absence of BMF in the patient with biallelic mutation of XRCC2, and the phenotypes of his cells, our data suggest that XRCC2 belongs to a subset of FA genes that are atypical and which function downstream in the FA–BRCA pathway.
Materials and methods
Additional details on Materials and Methods are provided as online supplementary information.
Supplemental material
Cell culture
Cells were grown as described previously,21 except where noted otherwise.
Cloning and mutagenesis
The R215X mutant of XRCC2 was generated using the QuikChange II Site-Directed Mutagenesis kit (Stratagene).21
Transfection and viral transduction
900677AT cells were reconstituted with XRCC2 using a retroviral vector, as described previously.9 ,18 For experiments involving the R215X mutant of XRCC2, cells were retrovirally transduced with the pOZ vector as described.22
Immunofluorescence microscopy
Cells were grown on coverslips, fixed, permeabilised, washed, incubated with primary and secondary antibodies, and mounted, as described previously.23
Microscopy, collection of images, counting of three replicates per sample and the generation of figures were as described previously.23
Immunoprecipitation
Cell lysis and immunoprecipitation assays, performed with anti-Flag M2 Affinity Gel (Sigma) or specified antibodies, were as described previously.21
Antibodies
Several commercially available primary antibodies were used: RAD51D (Novus), XRCC2 (Santa Cruz), BRCA1 (Millipore) and BRCA2 (Calbiochem). Anti-RAD51C, anti-XRCC3, anti-γH2AX, anti-haemagglutinin (HA) and anti-β actin antibodies were as described elsewhere.24
Secondary antibodies for immunofluorescence microscopy and the detection of immunoblots using chemiluminescence (Amersham) were as described previously.23
DNA damage sensitivity assays
For measurements of MMC, olaparib and formaldehyde sensitivity, cells were treated with MMC at doses ranging from 0 to 400 nM, olaparib at doses from 0 to 4 μM and formaldehyde at doses from 0 to 80 μM. Relative survival was measured using a colorimetric assay as described.21
For measurements of sensitivity to ionising radiation (IR), colony assays were performed on 900677AT cells, with or without correction with XRCC2, as described previously.24
Chromosome breakage analysis
For assays of chromosome breakage in primary and Lg T-immortalised lines, cells were treated with DEB and MMC, respectively. Cells were trypsinised, washed, swollen, fixed and dropped onto slides.25
The total number of aberrations per chromosome, including breaks, gaps and radials was counted as described,26 and the percentage of cells with one or more radial chromosomes was also calculated.
Results
Previously, we described a Saudi child born to healthy first cousin parents with a biallelic stop gain mutation in XRCC2 (transcript NM_005431), c.643C>T, predicted to encode p.R215*.20 This child showed intrauterine growth retardation, absent thumbs and radii, microcephaly, kidney malformations and other clinical features that are frequently observed in patients with FA.20 While these findings suggested the possibility that the patient had FA, mutations in other genes were also found. Thus, whether or not XRCC2 is a FA gene was not determined.
At the time of this current report, the boy is 7 years old, shows no signs of BMF and has normal haematological counts: haemoglobin 12.7 mg/dL, thrombocytes 397 000/μL, leukocytes 7000/μL and an absolute neutrophil count of 2100/μL.
To better understand the defect in 900677A cells, we quantified chromosome abnormalities induced by DEB in primary skin-derived fibroblasts from the patient in comparison to other primary fibroblast cell lines. The results (table 1) demonstrate that 900677A cells possess the characteristic hypersensitivity of FA cells towards DEB when compared with non-diseased normal fibroblasts (NL143889), similar to what has also been reported for RAD51C−/− cells.18 However, when compared with FANCA−/− (GM16631) and FANCC−/− (GM16754) fibroblast cells with defects in ‘early’ FA genes, the 900677A fibroblasts were less sensitive to crosslinkers. There was also an increased level of spontaneous chromosome breakage in 900677A cells, as compared with the normal fibroblasts, similar to that observed in FANCA−/− and FANCC−/− cells.
Spontaneous and DEB-induced chromosome breaks in 900677A and other lines
Next, to better understand the pathogenicity of the XRCC2 mutation, we assessed the levels of XRCC2 protein in SV40 large T-transformed 900677A (900677AT) cells. Although the c.643C>T mutation in XRCC2 is predicted to encode a mutant XRCC2 protein truncated at position R215 (p.R215X), we could not identify any XRCC2 protein band by western blot using an antibody directed against the N-terminus of XRCC2 (figure 1A), neither full-length (34 kDa) nor truncated XRCC2 (23 kDa). By comparison, full-length XRCC2 was readily detected in T-antigen transformed HEK293 cells.
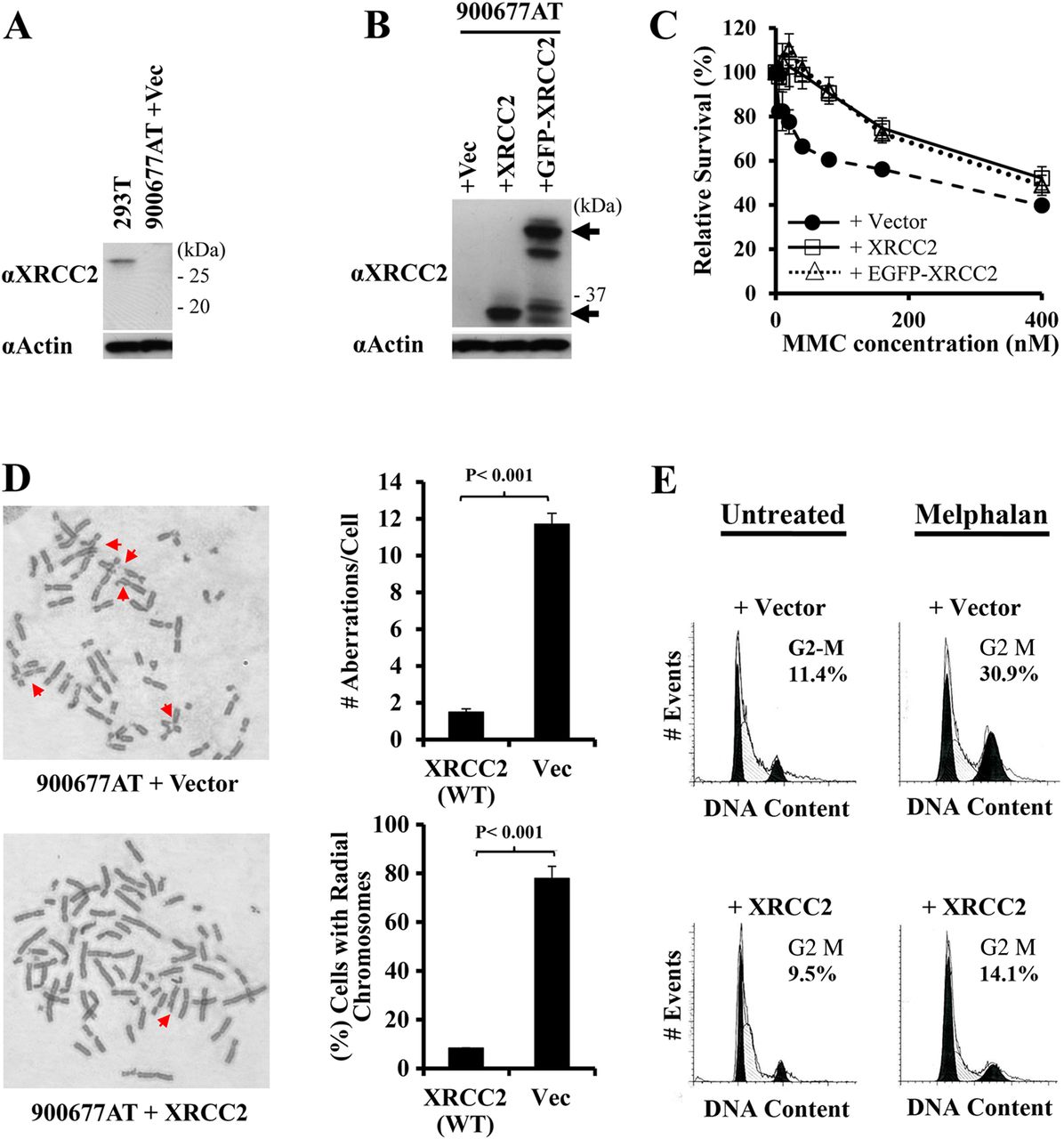
Complementation with XRCC2 corrects cellular phenotypes associated with sensitivity to DNA interstrand crosslinking agents that are characteristic of Fanconi anaemia cells. (A) XRCC2 was detected in extracts from a non-diseased individual (293T), but neither full-length (∼34 kDa) nor a truncated form of XRCC2 (expected at ∼23 kDa) was detected in extracts from 900677AT cells containing vector alone. (B) Retroviral expression of XRCC2, or a green fluorescent protein (GFP)–XRCC2 fusion protein, restored XRCC2 in 900677AT fibroblasts. Arrows indicate the position of the GFP–XRCC2 (above) and XRCC2 (below) proteins on immunoblots. Some degradation of GFP–XRCC2 is evident. Actin is shown as a loading control. Molecular weight markers are indicated in (A and B). (C) Relative survival of 900677AT fibroblasts immortalised with SV40 large T antigen, and which contained the lentiviral vector, XRCC2, or GFP–XRCC2, following treatment with a range of concentrations of mitomycin C (MMC). Values were normalised to levels in untreated populations for each cell line. (D) Examples are shown of chromosome aberrations in 900677AT immortalised fibroblasts reconstituted with vector or with XRCC2 following treatment with 300 nM MMC for 72 h (left). The total number of aberrations/metaphase cell, including breaks, gaps, radial and ring chromosomes, and the percentage of metaphase cells with one or more radial chromosomes, is shown for each cell line (right). (E) Histograms showing the cell-cycle distribution in 900677AT cells, with or without correction for XRCC2, in untreated populations or following treatment with 0.35 μg/mL melphalan. Curve fits for the G1 peak (left) and G2–M peak (right) were determined using ModFit and are shown in black in each histogram. The percentage of cells in G2–M is indicated on each histogram.
Re-introduction of XRCC2 corrects all FA-associated cellular defects in 900677AT cells
In 2000, Joenje et al28 proposed more stringent criteria to define a new FA gene: either correction of FA-related cellular phenotypes following exogenous expression of the candidate FA gene/cDNA or identification of two or more unrelated patients with biallelic mutations in the same gene. Given that only a single patient in the Saudi family described above has been identified so far worldwide as having biallelic mutations in XRCC2, we focused on functional genetic correction of the 900677A cells. To this end, we expressed the wild-type (WT) XRCC2 cDNA or a 5′ green fluorescent protein (GFP) fusion with XRCC2 in 900677AT cells using retroviral vectors. Figure 1B shows that the retroviral expression vectors introduced XRCC2 and also GFP-tagged XRCC2 into cells, while no full-length XRCC2 protein was detectable in control 900677AT fibroblasts transduced with the ‘empty’ vector (vec).
We then examined transduced 900677AT fibroblasts for all three cellular phenotypes that are typical of FA cells: sensitivity to DNA ICL agents, ICL-induced chromosome instability and ICL-induced accumulation in G2–M of the cell cycle.5 ,6 First, we demonstrate that expression of XRCC2, either with or without N-terminal GFP, corrected the cellular sensitivity to MMC (figure 1C). Next, we show that expression of WT XRCC2 in 900677AT cells dramatically reduced chromosome instability, as compared with 900677AT cells containing the empty vector. This is seen both in the examples shown and by quantification of chromosomal abnormalities using two distinct measures (figure 1D). Re-expression of XRCC2 corrected: (1) the total number of aberrations/cell, including breaks, gaps and radial chromosomes, and (2) the percentage of cells with radial chromosomes. Additionally, the DNA interstrand crosslinker melphalan induced accumulation in G2–M of 900677AT cells that contained the vector alone, but this was corrected by re-expression of XRCC2 (figure 1E). In summary, by each of three cellular phenotypes, 900677AT cells displayed a defective response to DNA ICL agents that is characteristic of FA. Importantly, in each case this pathological cellular phenotype was significantly corrected/complemented by expressing XRCC2 in the cells.
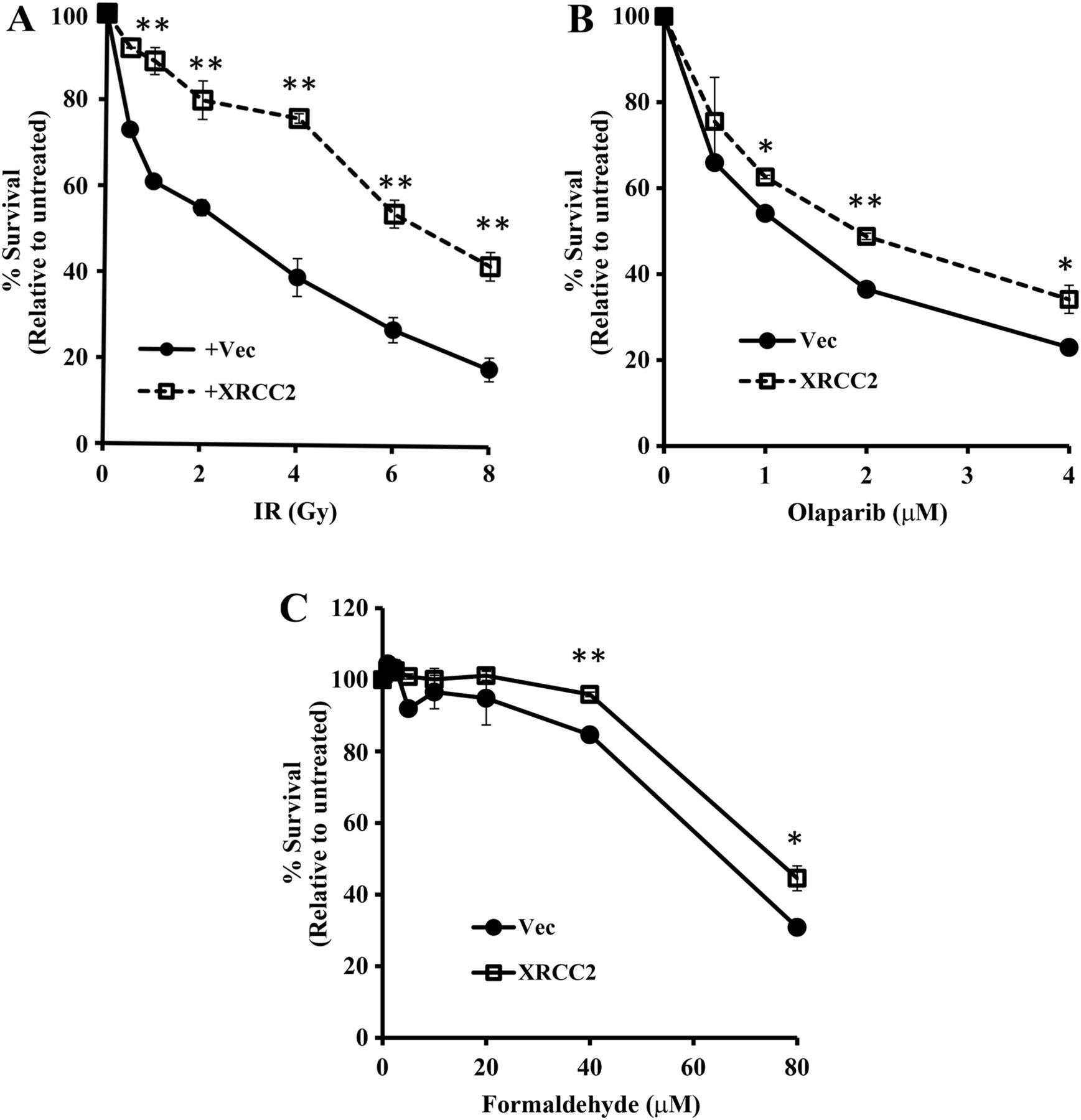
Next, to better understand the phenotype of human cells with a genetic deficiency for XRCC2, we examined the sensitivity of 900677AT cells to other DNA damaging agents (figure 2). Non-corrected 900677AT cells were particularly sensitive to IR, as compared with cells in which XRCC2 had been reintroduced (figure 2A). Interestingly, while upstream FA proteins have a minimal role in mediating resistance to IR,29 downstream FA proteins such as PALB2 and BRCA2 have a clear role.24 ,30 ,31

XRCC2 promotes cellular resistance to multiple DNA damaging agents, in addition to mitomycin C. Resistance of XRCC2−/− 900677AT cells, with or without reconstitution of XRCC2, to a range of doses of IR (A), the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib (B) and formaldehyde (C). Values for each cell line and type of DNA damage are relative to untreated populations for each cell line and that particular treatment. p Values, as determined using Student's t-test: *p<0.01, **p<0.0001.
The XRCC2 deficiency of 90677AT cells was also associated with increased sensitivity to the poly(ADP-ribose) polymerase (PARP) inhibitor olaparib (figure 2B) and to formaldehyde (figure 2C). This is consistent with previous reports that cells from certain patients with FA display increased sensitivity to either agent.10 ,24 ,31–33
In summary, these studies clearly demonstrate that the biallelic mutation in XRCC2, c.643C>T, which encodes p.R215X, is responsible for the FA-associated cellular defects in 900677A cells, as reintroduction of the WT protein corrects all pathological cellular phenotypes.
The R215X mutant of XRCC2 found in a patient with FA is unstable
Since the R215X mutant of XRCC2 was not detected in 900677AT cells (figure 1A,B), we assessed whether we could detect the truncated protein following stable expression in U2OS-DR cells along with a N-terminal Flag-HA epitope tag (figure 3A). While truncated XRCC2 was clearly present, based upon faster migration, the levels were dramatically lower than for the XRCC2 WT protein. To test the hypothesis that the p.R215X mutant of XRCC2 found in a patient with FA is unstable, we treated U2OS-DR cells expressing either the XRCC2 WT or R215X proteins with cycloheximide to inhibit new translation. As shown both on immunoblots (figure 3B) and on plots of protein band intensity (figure 3C), relative levels of the mutant XRCC2 R215X protein decreased more rapidly than the levels of WT XRCC2, thereby demonstrating that the XRCC2 R215X protein is indeed unstable with a shorter half-life. As further support for the increased turnover of R215X mutant protein, we show that inhibition of proteasome-mediated protein degradation with the MG132 inhibitor had a greater effect on levels of the XRCC2 R215X mutant than on levels of the WT protein, both on immunoblots (figure 3D) and in plots of band intensities (figure 3E). Thus, p.R215X may be pathogenic because of its relative instability.

The R215X truncation mutant of XRCC2, which is found in a patient with Fanconi anaemia (FA), is unstable. (A) As compared with wild-type (WT) XRCC2, the R215X truncation mutant of XRCC2, found in a patient with FA is expressed at low levels in U2OS-DR cells. (B) The R215X mutant of XRCC2 turns over more rapidly than the WT form, as shown by treatment with cycloheximide (CHX). Given the low levels of XRCC2–R215X, long exposures (long exp.) are shown for comparison to the WT form. (C) Signal intensity after treatment with CHX is shown for the WT and R215X forms of XRCC2 relative to the untreated lane. (D) Inhibition of proteasome-mediated protein degradation in U2OS-DR cells with MG132 has a greater effect on the R215X form of XRCC2 than on the WT. (E) Quantification of the effect of MG132 on the levels of WT and R215X XRCC2 relative to the levels at 4 h of treatment. Flag-HA-XRCC2 was used throughout the figure and actin is shown as a loading control.
The role/position of XRCC2 in the FA–BRCA pathway
Post-translational modification of FANCD2 by monoubiquitination is the central step in the FA pathway and is dependent on the normal function of ‘early’ FA gene products.7–9 ,14 Further, the monoubiquitination of FANCD2 is required for the assembly of FANCD2 into nuclear foci.14 Thus, as a first step towards determining the position of XRCC2 in the FA–BRCA pathway, we examined the levels of FANCD2 monoubiquitination and foci formation in 900677AT cells with or without re-expression of WT XRCC2 protein (see online supplementary figure S1). Monoubiquitinated FANCD2 was detected as a slower migrating band on immunoblots in untreated samples in both corrected and uncorrected 900677AT cells. Further, increased levels of monoubiquitinated FANCD2 were induced by MMC in 900677AT cells regardless of the status of XRCC2. Additionally, the assembly of FANCD2 foci in response to MMC was unchanged by proficiency or deficiency for XRCC2 (see online supplementary figure S1B,C). Thus, different from FA core complex components, and FANCD2 and FANCI, XRCC2 does not function early in the FA–BRCA pathway.
Supplementary figures
To determine whether XRCC2 may instead function as a late FA protein, we then examined RAD51 foci in 900677AT cells that were defective for XRCC2 (figure 4). As demonstrated in the example shown (figure 4A) and by quantification (figure 4B), XRCC2−/− fibroblasts had a pronounced defect in the assembly of RAD51 foci, either in untreated populations or following exposure to MMC. This defect was correctable by complementation with XRCC2. In contrast, in the examples shown in figure 4A, the assembly of γH2AX foci in response to MMC was similar in 900677AT cells either with or without restoration of XRCC2. Thus, 90677AT cells have a specific defect in the assembly of RAD51 foci.

XRCC2 promotes the assembly of RAD51 foci similar to other late Fanconi anaemia proteins. (A) Representative images of RAD51 foci detected in XRCC2−/− 900677AT cells, with or without reconstitution with XRCC2, using indirect immunofluorescence microscopy, following treatment with 0.5 μM mitomycin C (MMC) for 16 h. γH2AX foci demonstrate the assembly of DNA damage foci in both cell lines irrespective of the status of XRCC2. (B) Quantification of RAD51 foci in XRCC2−/− 900677AT cells, with or without reconstitution of XRCC2, either in untreated populations or following exposure to MMC.
Reconstitution of 900677AT cells with XRCC2 restored RAD51 foci to levels similar to those seen in fibroblasts from a control, WT individual (see online supplementary figure S2). Importantly, while early proteins in the FA pathway have a minimal role in promoting the assembly of RAD51 foci, cells with loss-of-function mutations in late FA genes, such as PALB2 and BRCA2, display marked deficiencies,19 ,34 as do 900677AT cells. Thus, taken together, our results strengthen the hypothesis that XRCC2 functions in the FA–BRCA pathway downstream of monoubiquitinated FANCD2 as a ‘late’ FA protein. The hypersensitivity of 900677AT cells to IR (figure 2A) is also consistent with this conclusion.
The interaction of XRCC2 with RAD51C, which is also a late and atypical FA protein, is missing in 900677AT cells
To better understand the function of XRCC2 as a FA protein, we sought to determine whether its interaction with another FA protein, RAD51C/FANCO,18 is disrupted in 900677AT cells (figure 5). This hypothesis was based on the fact that XRCC2 and RAD51C are both RAD51 paralogs which are known to interact.35 ,36 XRCC2 and RAD51C also display further similarity by having a common role in promoting the assembly of RAD51 foci.37 Consistent with previous reports,35 ,36 we found that RAD51C coimmunoprecipitated with XRCC2 in extracts from 900677AT cells that had been corrected by reintroduction of XRCC2 (figure 5A). As shown in figure 5B, the consequence of the c.643C>T mutation in 900677AT cells, which leads to undetectable XRCC2 protein, is that interaction with RAD51C cannot occur. As noted above (figure 5A), the XRCC2–RAD51C interaction was restored by reintroduction of XRCC2. Strikingly, we also found that RAD51C protein levels, while clearly detectable, were markedly decreased in XRCC2-deficient 900677AT cells, as compared with their genetically corrected counterparts (figure 5B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
XRCC2 maintains the levels of other proteins in the BCDX2 complex, and 900677AT cells have a diminished interaction of the XRCC2 and RAD51C Fanconi anaemia proteins. (A) The interaction of RAD51C with XRCC2 was detected in immunoprecipitation reactions performed using antibodies against XRCC2 or a non-specific control (IgG) with extracts derived from 900677AT cells in which XRCC2 had been reintroduced. (B) Levels of RAD51C that immunoprecipitated with XRCC2 using anti-XRCC2 antibodies in extracts from untreated XRCC2 proficient (XRCC2) or deficient (Vector) 900677AT cells. (C) The levels of various DNA damage response proteins, including each of the somatic RAD51 paralogs, in XRCC2−/− 900677AT cells either with or without exogenous expression of XRCC2. Cells were either left untreated or were incubated with 0.5 μM mitomycin C for 16 h, and each protein was detected using the indicated antibody. Actin is shown as a loading control for the levels of proteins present in extracts (B and C).
Given that RAD51B-C-D and XRCC2 form a complex together,35 ,36 we then determined the effect of mutation of XRCC2 in 900677AT FA cells on these other RAD51 paralogs (figure 5C). Importantly, each component of the BCDX2 complex was dramatically reduced in uncorrected cells, either with or without treatment with MMC, and XRCC2 and RAD51D were absent (figure 5C). Reconstitution of 900677AT cells with XRCC2 restored the levels of interacting RAD51 paralogs, such as RAD51B and RAD51C, to similar levels as in a normal fibroblast control, GM0637 (see online supplementary figure S3). Of note, RAD51C but not XRCC2 can form a separate complex with another RAD51 paralog, XRCC3.35 ,36 Proficiency or deficiency for XRCC2 had no clear effect on the levels of XRCC3, or on the levels of two other DNA double-strand break (DSB) repair-related FA proteins, RAD51 and BRCA1 (figure 5C). In summary, XRCC2 appears to have a specific function in maintaining the BCDX2 complex, which includes another late and atypical FA protein, RAD51C.
Discussion
Here, we identify XRCC2 as the 20th FA gene, which should therefore be labelled as FANCU. In fact, XRCC2 perfectly fits the profile of an atypical late FA gene as will be summarised below. Historically, prior to the discovery of any FA genes, patients with FA were defined clinically as presenting with the classical triad of congenital abnormalities, BMF and malignancies.38 Cells from these patients also displayed characteristic chromosomal breakage and hypersensitivity in response to DNA ICL agents.1 ,5 ,6 Subsequently, genes with loss-of-function mutations in patients with FA that presented with the triad described above were labelled as ‘FA genes’ since they complemented the cellular FA phenotype, specifically hypersensitivity to ICLs. However, in the last few years, biallelic or monoallelic germ-line defects have been identified in patients with cellular ICL hypersensitivity and increased chromosomal breakage that do not show the complete triad of clinical features, which are classically associated with FA. Patients with defects in these atypical FA genes, also called FA-like genes, including BRCA1/FANCS, RAD51/FANCR and RAD51C/FANCO, and as we show here, XRCC2/FANCU, have congenital abnormalities characteristic of FA but do not develop BMF.10 ,11 ,18
Each gene that is associated with FA, whether it be typical or atypical FA, encodes a protein that has an essential function in an elaborate mechanism that repairs DNA ICLs. This is shown schematically in online supplementary figure S4. Prominent steps in the repair of ICLs include recognition of the crosslink, incision to unhook the lesion, translesion synthesis to restore duplex DNA and HR to restart the replication fork.2 ,6 Proteins of the FA core complex and UBE2T/FANCT are involved early in the repair process, and are required for the assembly of FANCD2 and FANCI into nuclear foci.14–16 In contrast, homologous recombination occurs at later steps in the repair of ICLs and uses the products of other FA genes, such as BRCA1/2, PALB2 and RAD51. Related to their function in HR, these proteins are instead involved in the assembly of the RAD51 recombinase into DNA damage foci and are also required for resistance to IR.39 Thus, defects in the assembly of RAD51 foci observed in 900677AT cells position XRCC2 as a late FA protein.
Given the heterogeneity in clinical phenotypes,3 ,4 the defining features that are common to all FA complementations groups are cellular hypersensitivity to DNA ICL agents and the fact that this can be corrected by re-expression of the appropriate gene/protein.2 ,5 ,6 Ultimately, this hypersensitivity to ICLs reflects the common function for FA proteins in repairing this type of DNA lesion.
Bialleleic mutation of XRCC2 is responsible for all phenotypes in the patient's cells
XRCC2 is a bona fide FA gene by the accepted standard28 of correcting three different phenotypes of 900677A cells that are related to cellular sensitivity to ICLs. Further, XRCC2-deficient 900677A fibroblasts also have a modest increase in sensitivity to olaparib and formaldehyde. Increased sensitivity to PARP inhibitors, including olaparib, has been observed for FA cells with inactivation of other late FA genes, especially BRCA1/2, PALB2 and RAD51C.10 ,24 ,31 Notably, the increased sensitivity of 900677AT cells to formaldehyde, which can be corrected by re-expression of XRCC2, is also consistent with our having identified them as FA cells. Sensitivity to formaldehyde and other aldehydes has been observed in a variety of cell lines with deficiencies in FA proteins.32 ,33 ,40–43
900677A cells, with or without complementation by XRCC2, display no defect in FANCD2 monoubiquitination or foci formation. Since monoubiquitination of FANCI by the FA core complex is essential for FANCD2 monoubiquitination and assembly into foci,15 ,16 FANCD2 is a reliable indicator that XRCC2 does not function early in the FA–BRCA pathway, but instead functions late. In support of this conclusion, XRCC2-deficient 900677AT cells display cellular hypersensitivity to either ICLs and IR, similar to cells with deficiencies for other late FA genes. Importantly, these cellular phenotypes are correctable by re-expression of XRCC2.
XRCC2 interacts with and stabilises RAD51B, RAD51C/FANCO and RAD51D but not XRCC3
The mutant R215X protein is detected at decreased levels as compared with the full-length protein. This appears to result from protein instability rather than non-sense-mediated decay of mRNA, since the truncating mutation is in the last exon of XRCC2.44
Given their deficiency for XRCC2, 900677AT cells lack the interaction of XRCC2 with another FA protein, RAD51C. We also show, in figure 5, that all components of the BCDX2 complex, including RAD51C, are destabilised when WT XRCC2 is missing in 900677AT cells. These effects may explain the pathogenicity of the p.R215X mutant.
Our hypothesis is that the deficiency of 900677AT cells for XRCC2 results in the destabilisation of RAD51D, since these proteins directly interact.45 The fact that RNAi-mediated depletion of the XRCC2 protein results in dramatically decreased levels of RAD51D is consistent with this hypothesis.46 Instability of RAD51D then likely leads to destabilisation of its direct binding partner, RAD51C.45 Destabilised RAD51C could, in turn, lead to instability of RAD51B, since these proteins also directly interact.45 Consistent with a primary effect of XRCC2 deficiency on RAD51D stability, RAD51D was absent in 900677AT cells, in contrast to RAD51B and RAD51C, which were still detectable, though at lower levels (figure 5).
Taken together, the association of XRCC2 with FA suggests the possibility that a key function of the XRCC2 protein in the FA–BRCA pathway is to stabilise the other members of the BCDX2 complex, all of which function in HR.37 Interestingly, since RAD51D encodes a protein that directly interacts with the two other RAD51 paralogs that are FA proteins, RAD51C/FANCO and XRCC2/FANCU, RAD51D appears to be an important candidate FA gene.
Atypical FA genes
As our XRCC2 patient is still younger than the median age of onset of BMF in patients with FA, which is 7.6 years of age,3 the vast majority of whom have defects in early FA genes, it is possible that haematopoietic defects will simply arise later. Also, it should be noted that while the patient initially displayed increased chromosome breakage in blood cells,20 tests to exclude somatic reversion as an explanation for the absence of haematologic phenotypes have not been performed recently.
All the same, loss-of-function mutations in BRCA1, RAD51, RAD51C and now XRCC2, all of which are late FA genes, have not been associated with BMF.10 ,11 ,18 On this basis, we designate these genes, including XRCC2, as atypical FA genes. In this context, it should be noted that BMF is not even observed in 100% of ‘typical’ FA patients with mutations in early FA genes.3
Our classification of XRCC2 as an atypical FA gene advances the understanding of the FA–BRCA pathway in multiple ways. First, our results have yielded a more complete knowledge of genes that cause FA when mutated. Second, these results further suggest that atypical FA genes, including BRCA1/FANCS, RAD51C/FANCO, RAD51/FANCR and now XRCC2/FANCU, encode proteins that function late in ICL repair. Members of this subgroup of FA genes are defined by a common role in regulating RAD51 foci, HR and resistance to IR.10 ,18 ,37 ,47
The fact that heterozygous mutation of XRCC2 is also associated with a predisposition to breast cancer48 is consistent with the conclusion that XRCC2 is an atypical late FA gene. Heterozygosity for two other atypical, and late, FA genes, BRCA1 and RAD51C, is also associated with an increased risk of developing breast cancer, unlike heterozygosity for early FA genes.48–51
Acknowledgments
We are grateful to Dr. James Lessard (Cincinnati Children's Research Foundation) and Dr. Yoshihiro Nakatani (Dana-Farber Cancer Institute) for anti-actin antibodies and pOZ vectors, respectively. We thank Fan Zhang, Christophe C. Marchal, Stephanie L. Kelich and Felicia M. Kennedy for expert technical assistance.
References
Footnotes
HH and PRA made equal contributions to this study.
Contributors J-YP, ELV, AJ, CW, SCC and GHV performed the studies. MO was responsible for clinical care. FSA provided cell lines. J-YP, PRA and HH were responsible for study design. PRA, J-YP and HH wrote the paper. CW and FSA edited the paper.
Funding This work was supported by National Institutes of Health (grant numbers R01 CA155294 to HH and R01 HL085587 to PRA), and also the Lilly Foundation Physician/Scientist initiative and the Walther Cancer Foundation to HH.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All data relevant to this study have been included in the manuscript and supplemental materials.