Article Text

Abstract
Background Ritscher–Schinzel syndrome (RSS) is a clinically heterogeneous disorder characterised by distinctive craniofacial features in addition to cerebellar and cardiac anomalies. It has been described in different populations and is presumed to follow autosomal recessive inheritance. In an effort to identify the underlying genetic cause of RSS, affected individuals from a First Nations (FN) community in northern Manitoba, Canada, were enrolled in this study.
Methods Homozygosity mapping by SNP array and Sanger sequencing of the candidate genes in a 1Mb interval on chromosome 8q24.13 were performed on genomic DNA from eight FN RSS patients, eight of their parents and five unaffected individuals (control subjects) from this geographic isolate.
Results All eight patients were homozygous for a novel splice site mutation in KIAA0196. RNA analysis revealed an approximate eightfold reduction in the relative amount of a KIAA0196 transcript lacking exon 27. A 60% reduction in the amount of strumpellin protein was observed on western blot.
Conclusions We have identified a mutation in KIAA0196 as the cause of the form of RSS characterised in our cohort. The ubiquitous expression and highly conserved nature of strumpellin, the product of KIAA0196, is consistent with the complex and multisystem nature of this disorder.
- Clinical genetics
Statistics from Altmetric.com
In 1987, a new and presumed autosomal recessive malformation syndrome was described in two Caucasian sisters who displayed distinctive craniofacial features (macrocephaly, prominent forehead, hypertelorism, downslanting palpebral fissures, depressed nasal bridge and low-set ears), cerebellar anomalies, congenital heart defects and intellectual disability.1 Since the initial report, sporadic and familial cases of what is now named Ritscher–Schinzel syndrome (RSS), or 3C (cranio-cerebello-cardiac) syndrome (MIM 220210), can be found in the literature.2–12 Because RSS/3C has been described in individuals from different populations, and because phenotypic variability (even among affected siblings) is well documented, Leonardi et al8 attempted to define minimum diagnostic criteria by reviewing 28 selected RSS cases. Even though cardiac (primarily septal defects) and cerebellar anomalies (primarily Dandy–Walker malformation or Dandy–Walker variant) occurred in approximately 80% of patients, Leonardi et al8 concluded that the pattern of craniofacial features can be an important diagnostic tool, a sentiment recently echoed by Craft et al.10
We have been following a large cohort of First Nations (FN) patients (initially described by Marles et al)5 who uniformly display prominent forehead, low-set ears, hypertelorism and wide, downslanting palpebral fissures. They also show brachycephaly, low posterior hairline and intellectual disability (figure 1 and see online supplementary table S1). Variable phenotypic traits among the members of our cohort include atrial and ventricular septal defects, Dandy–Walker malformation or Dandy–Walker variant and ocular coloboma. In spite of variable cerebellar and cardiac involvement, our cohort originates from an isolated, remote community in northern Manitoba, Canada, allowing for investigations to identify the underlying shared genetic defect.
Genomic DNA from eight FN patients, eight of their parents and five unaffected individuals (control subjects) from the same geographic region was sent to The Centre for Applied Genomics (The Hospital for Sick Children, Toronto, Ontario). Genotypes for SNPs of the Affymetrix Genome Wide Human 6.0 Array were generated, and all data was analysed for loss of heterozygosity using Genotyping console 4.1. The only identically homozygous region (size range from 1–20Mb) shared by all eight affected individuals was a nearly 1Mb region on chromosome 8q24.13 from rs6470263 to rs1580096, a 956,528 bp region (GRCh37/hg19 assembly). None of the parents or the five control subjects were homozygous for the entire interval. Because patients with a submicroscopic 6p25 deletion 13–15 have a similar phenotype to RSS patients, we evaluated copy number (CN) variation. None of the eight patients showed a CN loss at 6p25, nor did they share a CN gain or loss in any other chromosomal segment, including the identified homozygous interval.
The 8q24.13 candidate gene region contained six known genes (MTSS1, ZNF572, SQLE, KIAA0196, NSMCE2 and TRIB1), and all were Sanger sequenced (as described in online supplementary methods). The only biallelic mutations identified occurred in KIAA0196, where sequence analysis revealed homozygosity for three novel (not listed in dbSNP) variants at c.3335+2T>A, c.3335+4C>A and c.3335+8A>G in each patient (figure 2A). All parents were heterozygous for the three sequence changes, and none of the five control subjects was homozygous for any of these changes.

Consistent craniofacial features in our cohort including: prominent forehead, low-set ears, hypertelorism with wide, downslanting palpebral fissures, brachycephaly and low posterior hairline. Row 1—patient 3, row 2—patient 4, row 3—patient 5, row 4 (a-e) patient 7; (f-h) patient 10.

{kind=link}
{kind=link}
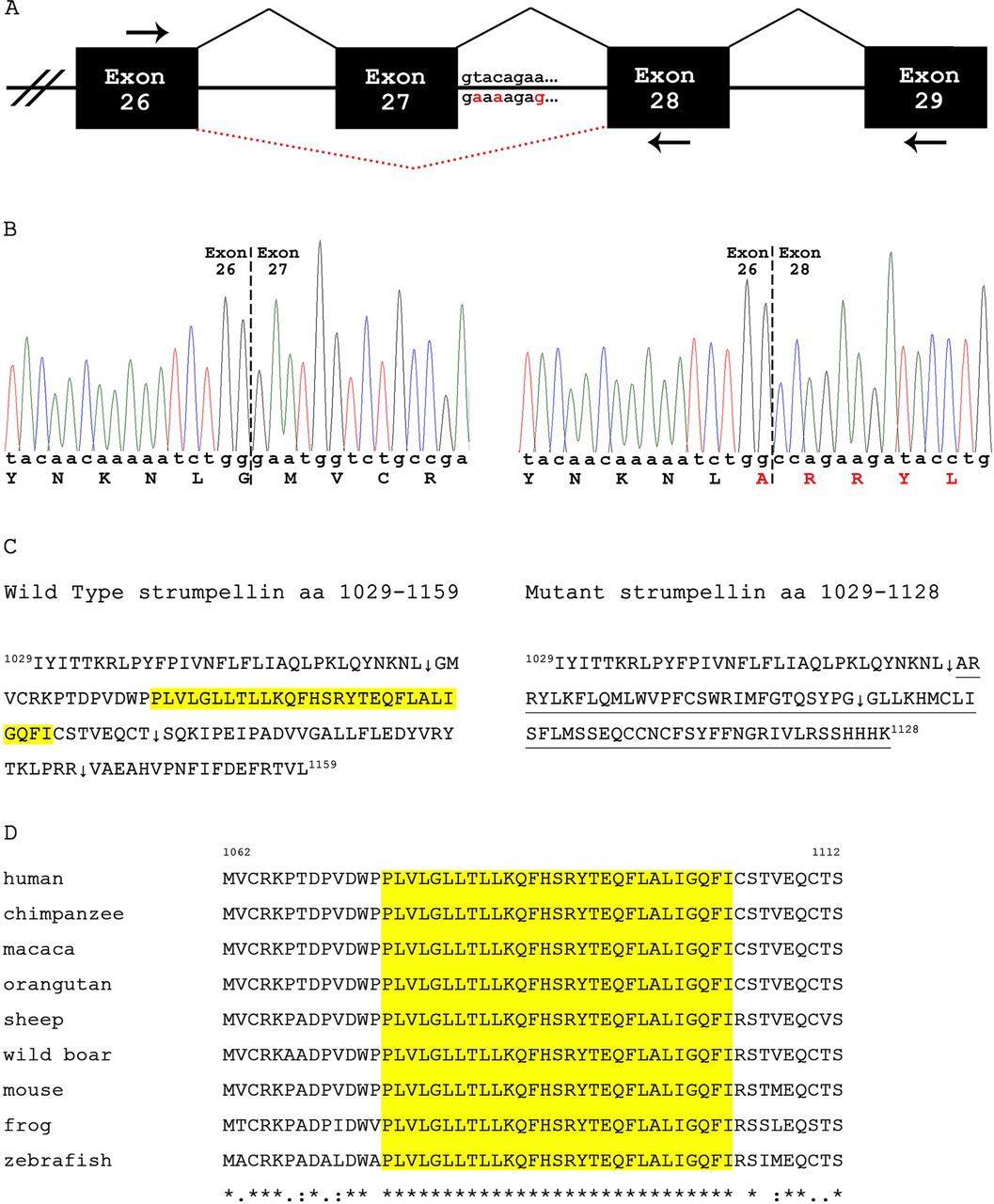
KIAA0196 exon 27 donor splice site mutation in First Nations (FN) patients. (A) Genomic organisation of exons 26 to 29. The c.3335+2T>A, c.3335+4C>A and c.3335+8A>G mutations are highlighted in red below the wild type intron 27 sequence. Dashed lines (in red) depict the aberrant splice product. (B) Partial sequence of control (left) and patient (right) cDNA. The Ritscher–Schinzel syndrome (RSS) mutation in KIAA0196 results in skipping of exon 27. (C) C-termini of the wild type (amino acids 1029–1159) and mutant (amino acids 1029–1128) strumpellin. Only wild type strumpellin possesses the 29 consecutive, highly conserved amino acids encoded by exon 27 (highlighted in yellow). Splicing exon 26 to exon 28 causes a frameshift: the novel 68 C-terminal amino acids are underlined. Arrows follow the last amino acid encoded by exons 26, 27 and 28 (wild type) and exons 26 and 28 (mutant). Normal and mutant strumpellin weighs 134.3 kDa and 131.1 kDa, respectively. (D) Sequence alignment of strumpellin amino acids encoded by exon 27. Probcons (v.1.09) was used for the cluster analysis. Highlighted (yellow) are the 29 consecutive amino acids that are completely conserved (from zebrafish to humans).
The c.3335+8A>G mutation created an AvaII restriction site allowing us to distinguish between the wild type (480 bp) and mutant (333 bp/147 bp) PCR alleles (see online supplementary methods, table S2) in a population survey (selected from the northern community of interest). Genomic DNA from 133 of 600 newborn blood spots collected over the past 2 years were anonymised and analysed for the c.3335+8A>G mutation of KIAA0196. Fifteen individuals were heterozygous for the mutation, meaning approximately one in nine individuals from the most recent generation of this isolate is a RSS carrier. This number predicts that in the next generation, approximately one in every 325 children born in this community will have RSS. Similar carrier frequencies due to founder mutations are not uncommon in genetic and geographic isolates. For example, the Limb Girdle Muscular Dystrophy mutation in TRIM32 (p.Asp487Asn) has an estimated frequency of 1 in 6.5 in Hutterite individuals.16 None of 212 chromosomes from 106 random Caucasian individuals carried the c.3335+8A>G mutation. Finally, using the same molecular test, we have been able to confirm the clinical diagnosis of RSS in three additional FN patients (not included in the SNP analysis), and have also determined that two of these patients are homozygous for all three KIAA0196 mutations. Clinical features of two (9 and 10) of these patients are included in online supplementary table S1, and are consistent with the other eight patients.
To assess the potential effect of the identified intronic mutations, we performed real-time RT-PCR using RNA prepared from whole blood from two patients and from a control subject (see online supplementary methods, table S2). Comparison of normalised cycle threshold (Ct) values indicated a 6.98 to 8.72 (mean 7.85)-fold reduction in the relative amount of KIAA0196 transcript in the patient samples versus the control sample. Sanger sequencing of the cloned PCR product from a patient revealed that the primary product did not contain exon 27 (figure 2B). Taken together, these results suggest that the major altered KIAA0196 transcript produced by the patient might be targeted for nonsense mediated decay.
NNSPLICE 0.9 (http://www.fruitfly.org/seq_tools/splice.html) analysis of the wild type and all combinations of the single, double or triple mutants revealed that only the c.3335+2T>A mutation reduced the donor splice site score below the default detection threshold. The presence of the c.3335+4C>A or c.3335+8A>G mutations, alone or in combination with each of the others had no effect.
Strumpellin, the product of KIAA0196, is a 134 kDa (1159 amino acid) glycoprotein that is highly conserved (from plants to humans) and ubiquitously expressed.17 It is predicted to have multiple transmembrane domains, with the N-terminal region (amino acids 1-240) comprising six α helices and two β strand segments, the central region (amino acids 241-971) comprising spectrin-repeat domains and the C-terminal region (residues 792-1159) showing structural similarity to exportin-5 and importin β-1 (molecules critical in the transport of microRNA precursors).17 Of the three base pair changes identified in our cohort, only c.3335+2T>A is predicted to have a functional impact on splicing. The major transcript skips exon 27 (figure 2A,B) causing a frameshift and a premature stop after amino acid 1128. Consequently, the mutant strumpellin is missing 99 C-terminal amino acids normally encoded by exons 27–29; these have been replaced by 68 novel C-terminal residues (figure 2C). Of note, the variant strumpellin is missing 29 consecutive amino acids that are completely conserved (from zebrafish to humans) encoded by exon 27 (figure 2D); the function of this region is currently unknown. Strumpellin western blot analysis from a patient sample indicated that the amount of strumpellin present in the RSS patient was reduced by approximately 60% as compared to control (see online supplementary methods, figure S1). In the absence of wild type transcripts in this patient, the protein detected is likely the 1128 amino acid mutant strumpellin (figure 2C).
A previous report identified three KIAA0196 missense mutations (c.1411A>G, N471D; c.1857G>C, L619F; c.1876G>T, V626F) underlying an autosomal dominant spastic paraplegia (SPG8).18 These variations all reside within the spectrin-repeat domains, but only amino acids 619 and 626 are highly conserved across species. Rescue studies in zebrafish showed that the 619F and 626F variants impaired normal protein function, whereas 471D did not.18 The reduced amount of strumpellin variant in our patients does not result in a progressive neuromuscular phenotype seen in patients with SPG8 likely due to the intact N-terminal and spectrin-repeat domains.
Although a strumpellin knockout model organism has not yet been characterised, strumpellin knockdown studies in zebrafish17 have a defined phenotype which includes tail curvature, impaired motility and cardiac oedema (leading to contractile dysfunction). The same investigators performing small hairpin RNA-mediated knockdown studies in human SH-SY5Y neuroblastoma cells observed decreased axonal length. Patients with RSS have intellectual disability and congenital malformations involving multiple systems. The ubiquitous expression and high conservation of strumpellin suggests that alterations in this protein, as predicted by the mutation identified in our cohort, may result in multiple clinical effects.
This report is the first to identify a specific gene (KIAA0196) involved in RSS/3C syndrome. Because of craniofacial similarities between our patients and those described by Khalifa et al9 and Craft et al,10 molecular analyses of KIAA0196 in their patients is indicated. The study of additional patients will help to elucidate the clinical and possible genetic variability of RSS.
Acknowledgments
We would like to thank the patients and family members for participating in this study, and the newborn screening section of Cadham Provincial Laboratory for providing the dried filter paper blood specimens. This study was funded by operating grants from the Manitoba Medical Service Foundation and the Manitoba Institute of Child Health (to AME) and the Winnipeg Rh Institute Foundation (to TZ). TB received a summer studentship from the Manitoba Institute of Child Health. GMH is a Canada Research Chair in Molecular Cardiolipin Metabolism.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement 1
- Data supplement 2 - Online figure
Footnotes
-
Contributors Phenotypes were determined by AEC, AME, BNC and CRG. TB was the study coordinator. GC performed the SNP analysis, VL the molecular analysis and GMH the protein analysis. AME, LRS and TZ designed the study, analysed the data and wrote the manuscript.
-
Funding Manitoba Medical Service Foundation, Manitoba Institute of Child Health.
-
Competing interests None.
-
Ethics approval University of Manitoba Research Ethics Board.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Patient consent Obtained.