Article Text

Abstract
Motor kinesins are a family of evolutionary conserved proteins involved in intracellular trafficking of various cargoes, first described in the context of axonal transport. They were discovered to have a key importance in cell-cycle dynamics and progression, including chromosomal condensation and alignment, spindle formation and cytokinesis, as well as ciliogenesis and cilia function. Recent evidence suggests that impairment of kinesins is associated with a variety of human diseases consistent with their functions and evolutionary conservation. Through the advent of gene identification using genome-wide sequencing approaches, their role in monogenic disorders now emerges, particularly for birth defects, in isolated as well as multiple congenital anomalies. We can observe recurrent phenotypical themes such as microcephaly, certain brain anomalies, and anomalies of the kidney and urinary tract, as well as syndromic phenotypes reminiscent of ciliopathies. Together with the molecular and functional data, we suggest understanding these ‘kinesinopathies’ as a recognisable entity with potential value for research approaches and clinical care.
- clinical genetics
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The mammalian kinesin superfamily proteins (KIFs) are microtubule and ATP-dependent molecular motors, which were first identified in 1985 as axonal transporters in squid and bovine brains.1 Forty-five different kinesin family member (KIF) genes were identified in the mouse genome so far, 44 of which are present in the human genome. Phylogenetic analysis based on sequence homology between the human and the mouse genome led to the classification of KIF genes into 16 families, from kinesin-1 to kinesin-14B (figure 1).2 The first kinesins discovered belong to the kinesin-1 family (KIF5A, KIF5B and KIF5C), and they form a heterotetramer of two heavy chains and two light chains (KLC1-4).2 KIF genes encode KIFs, a specific class of motor proteins generating intracellular motility by driving directional transport of various cargoes such as organelles, protein complexes and mRNAs along the microtubule system.2 Studies using knockout mouse models by Hirokawa and colleagues significantly contributed to elucidate the roles of kinesins in mammalian physiology. Their role in transport is fundamental to cellular logistics and performance, and the molecular motors are not only effectors of signal transduction cascades but also transport and/or bind to important signal transduction molecules to actively modulate their function.3

Phylogenetic tree of mammalian kinesin superfamily genes identified in the human (and mouse) genome and classified in 16 subfamilies (from kinesin 1 to 14B) (adapted from Hirokawa et al 3).
The first kinesins were observed in the context of axonal transport in neurons, and a novel disease entity of ‘motor–proteinopathy’ was proposed for the pathogenesis of axonal neuropathies in 2001.4 Due to their role in cellular membrane trafficking, however, kinesins are essential for the functioning of many polar cell types, such as neurons, epithelial cells, sperm cells or stem cells during organogenesis. Kinesins also play a fundamental role in cell-cycle dynamics, both during mitotic and meiotic processes. They regulate chromosomal condensation and alignment, spindle formation, cytokinesis and cell-cycle progression.5 It is estimated that about a dozen kinesins are involved in the cell cycle. Among these, there is a specific subclass of chromokinesins (kinesin 4 and kinesin 10 family) which are able to bind chromosomes.6 Recently, KIFs were discovered to act as microtubule stabilisers (KIF26A and KIF21A) and depolymerisers (KIF2A and KIF2C), activities which are important for both cellular morphogenesis and mammalian development, playing a role in neuronal and axonal morphology and ciliogenesis.7
Alterations in motor kinesins are leading to human disease by various pathological mechanisms, including cancer and multifactorial and monogenic disorders. Variants in 18 out of the 44 human KIF genes were identified to cause monogenic disorders, following different modes of Mendelian inheritance and associated with a wide spectrum of clinical signs. These range from lethal and multiple to isolated congenital anomalies—including birth defects potentially detectable in the foetal period by current prenatal imaging studies—to postnatally apparent neurodevelopmental disorders, intellectual disability and neurological conditions.
We will review the current state of knowledge of the biological processes kinesins are involved in and discuss their emerging role in human disease, particularly in birth defects and congenital anomaly syndromes. Birth defects remain a leading cause of perinatal lethality in industrialised countries.8 Structural anomalies are recognised with increasing reliability during early pregnancy by the use of high-resolution ultrasound technologies, thus raising questions about diagnosis, aetiology, prognosis and recurrence risk, particularly in the presence of more than one anomaly, which most likely indicates a genetic aetiology. We identify recurrent phenotype patterns caused by alterations in KIF genes, and we outline the complexity of phenotype–genotype correlations mirroring the processes of intracellular microtubule-mediated transport and movement, in which kinesins play a fundamental role. There are likely many more relationships between the clinical signs and the genetic variants to be identified in the future, and the functional network of kinesins and their role in human disease need to be further elucidated. We propose to introduce the term ‘kinesinopathies’ for this group of conditions, which are phenotypically and genetically overlapping and characterised by the functional impairment of a specific group of molecular motors. We hope that their systematic approach leads to a better recognition in clinical practice, as well as in genome-wide sequencing for diagnosis and research, and opens strategies for the future development of molecular therapies.
KIF structure
All KIFs have a phylogenetically well-conserved motor domain head, consisting of an ATP-binding motif and a microtubule-binding domain. Depending on the position of the motor domain, kinesins can be subdivided into N-kinesins (amino-terminal motor domain), M-kinesins (middle-region motor domain) and C-kinesins (carboxy-terminal motor domain).2 Most kinesins belong to the N-kinesin subgroup, but members of the kinesin 13A family (figure 1) belong to the M-kinesin subtype, while KIF1C, KIF2C and KIF3C belong to the C- kinesin subfamily.3 Both N-kinesins and C-kinesins are responsible for plus end and minus end-directed motility, M-kinesins for depolymerisation of microtubules in tubulin molecules. However, there are a few exceptions to this categorisation.9 The motor domain head attaches to the neck, the coiled coil stalk and the tail. The kinesins’ neck is family-specific and responsible for the direction of motility or regulation of activity. The coiled coil stalk and tail are involved in kinesin dimerisation and/or interactions with cargoes. Kinesins typically use scaffold proteins and adaptor proteins to bind their cargoes but can sometimes bind the cargo directly. Scaffolds and adaptors might also have regulatory roles in kinesin-driven intracellular transport, that is, recognising specific cargoes and regulating their loading and unloading.3
Role of KIFs in physiology and disease
The application of genome-wide sequencing for gene identification in research or for clinical diagnostic purposes significantly contributes to the identification of KIF candidate genes. Genotype–phenotype correlations in KIF gene-related disorders, together with functional and animal studies, continue to elucidate the complex involvement of KIFs in human developmental pathways and disease. Table 1 summarises the monogenic conditions caused by variants affecting the function of KIF genes.
Specific monogenic disorders caused by variants affecting the function of KIF genes
Summary of phenotypes and genotypes of KIF14 9 26 30 31
The kinesins’ functions in physiological processes, however, are complex and still incompletely understood, but their role in cell-cycle progression and regulation, including both meiosis and mitosis, in intracellular trafficking, axonal transport, microtubule activity and ciliogenesis, is increasingly studied. Figure 2 summarises the clustering of KIF genes according to their functional roles and the phenotypical consequences as identified to date in 32 out of the 44 human kinesin genes.
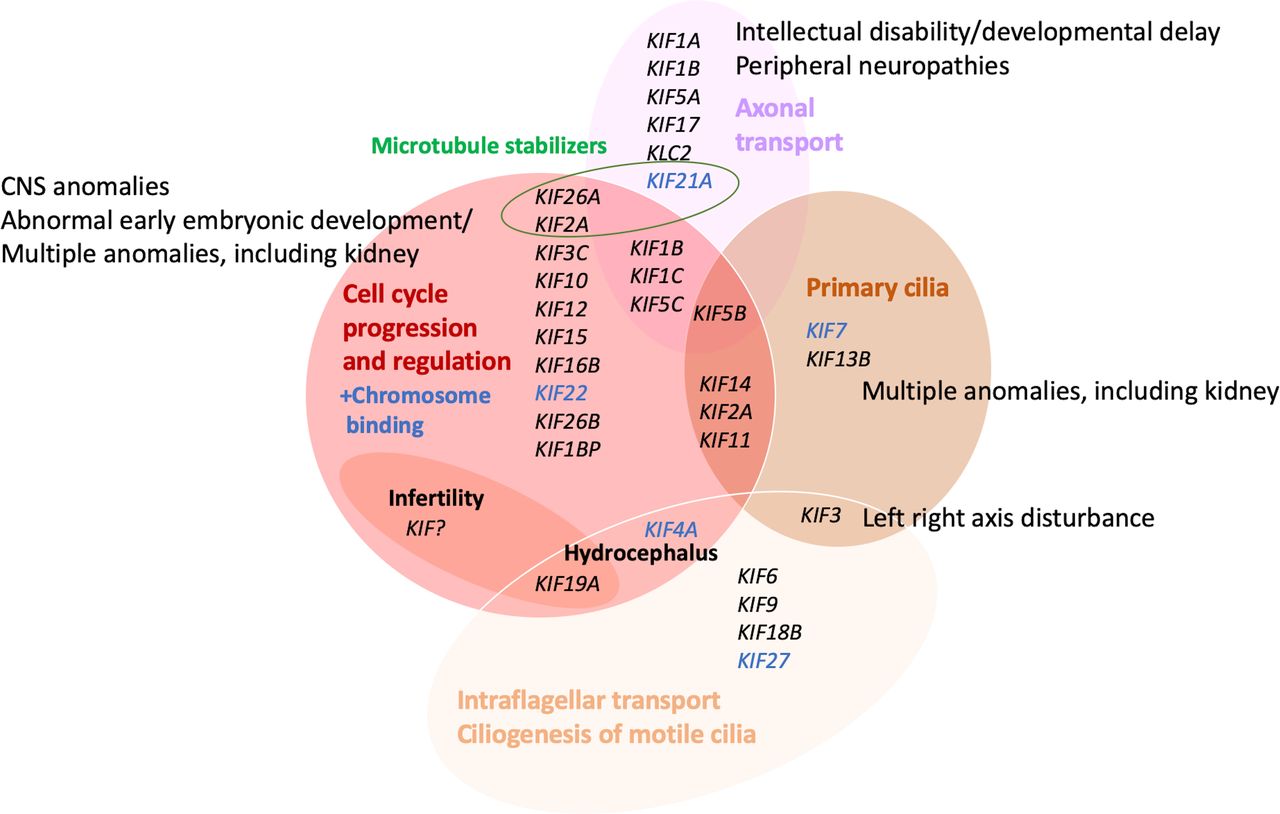
Assignment and clustering of KIF genes to various functions and relation to birth defect or monogenic phenotype groups. Detailed phenotypes are shown in tables 1 and 3. Cancer and multifactorial conditions are not included. CNS, central nervous system.
Kinesins play a pivotal role during early development and organogenesis. Microcephaly is one of the most frequently associated clinical signs, mirroring a defect in the regulation of the final number of neurons during development.10
KIF4A is a motor protein that translocates PRC1, a cytokinesis protein, to the ends of the spindle microtubules during mitosis, regulates the PARP1 activity in brain development and the survival of neurons, and is a member of the L1CAM recycling pathway. Variants in L1CAM cause X-linked isolated and syndromic hydrocephalus. KIF4A was recently proposed as a candidate gene for hydrocephalus.11
KIFs are involved in neuronal branching, and microtubule depolarisation, operated by KIF2A M-kinesin, was suggested to suppress collateral branch extension during brain development, leading to anomalies of cortical development, including agyria and pachygyria, subcortical band heterotopia and corpus callosum anomalies.12
Functional disruption of KIF genes in knockout mice often results in embryonic lethality, for example, for Kif18A, Kif10, Kif3A, Kif3B and Kif5B,13–17 highlighting the importance of kinesins in embryonic and foetal development. A study on KIF16B demonstrated that microtubule-based trafficking is responsible for early development and stem cell survival.18 KIF26B is essential in kidney development, contributing to the adhesion of mesenchymal cells to the ureteric bud.3 KIF26A was suggested to play a role in enteric nervous system development, because knockout mice develop a megacolon and enteric nerve hypoplasia,19 and to negatively regulate nociceptive sensation.20
A significant number of KIFs play a prominent role in ciliogenesis and cilia function. They regulate cilia length, ciliary assembly/disassembly and can have motile cilia-specific functions.21 Some KIFs, specifically found in primary cilia (PC), regulate the length of the axoneme and its disassembly when re-entering the cell cycle.
KIF7, also a key component of the Hedgehog signalling pathway, is responsible for cilia length regulation through suppression of microtubule polymerisation.7 KIF7 variants cause hydrolethalus, acrocallosal, and Joubert and Al-Gazali-Bakalinova syndromes.22 Kif2A knockout mice have severe brain defects, and KIF2A variants in humans lead to microcephaly because of cell-cycle delay in cellular progenitors resulting from cilia disassembly defects. KIF24, belonging to the same kinesin 13 family, plays a role in both microtubule depolymerising activity and regulation of the early steps of ciliogenesis. Other PC-related KIFs recently identified are KIF5B, KIF1C and KIF13B, and a potential role in cilia was hypothesised for KIF11 and KIF14.
KIF3 protein complex (KIF3A-KIF3B-KAP3 heterotetramer) is a molecular motor necessary for intraflagellar transport (IFT) but is also involved in ciliogenesis of motile cilia. Kif3a-knockout or Kif3b-knockout mice are prenatally lethal, exhibiting anomalies similar to ciliopathy phenotypes, including the disturbance of left–right body determination.3
KIF19A is localised at the tip of motile cilia and performs motor and microtubule-depolymerising activities during IFT. Kif19a-knockout mice present with hydrocephalus and female infertility, common signs in ciliary defects, due to abnormally elongated cilia with altered motility, not able to generate proper fluid flow.9
Further KIFs, which may have specific roles in motile cilia, are KIF27, KIF9, KIF6 and KIF18B. Regarding the involvement of numerous KIFs in cilia-related processes, it is not surprising that many disorders caused by variants affecting KIF gene function are presenting with anomalies reminiscent of ciliopathies.
Kinesin motors have a fundamental role in neuronal function, as they are responsible for the transport of synaptic vesicle precursors and transmitter receptors along axons and dendrites from the neuron body.3 Molecular motor activity as for KIF1A, KIF5 and KIF17 is important for higher brain functions, such as learning and memory through regulation of synaptic transmission.5 Dysfunction can be associated with intellectual disability and global developmental delay (table 1).
Impaired function can also result in peripheral neuropathies (KIF5A, KLC2, KIF1A and KIF1B) and ocular motility disorders (KLC2 and KIF21A)23 24 when axon elongation in the peripheral nervous system and optic nerve is affected. KIF5A variants are associated with epileptic phenotypes both in humans and mice25 because the transport of neurotransmitter receptors is disturbed and inhibitory regulation is altered.
Due to their role in cell-cycle regulation, kinesins are important in male spermatogenesis and female oogenesis. They are involved in all steps of spermatogenesis 26 and, based on previous animal studies, they may represent a potential target to treat male infertility. In female meiosis, 13 KIF genes were studied in animal models. There is some evidence that kinesin expression is vulnerable to maternal ageing and environmental factors, such as oocyte cryopreservation and alcohol consumption. It may be promising to expand research in this field in order to clarify the mechanisms and factors contributing to oocyte quality decline.27
Many kinesins were extensively studied in the fields of cancer development, progression and therapy. Deregulation of the mitotic kinesins by both overexpression and decreased expression causes cancer progression or can be a prognostic marker in various tumours.28 The cell-permeable small-molecule mitotic inhibitor monastrol was discovered in 199929 and was shown to arrest cells in mitosis by specifically inhibiting KIF11, a kinesin important for spindle bipolarity. The bipolar mitotic spindle is replaced by a monoastral microtubule array surrounded by a ring of chromosomes, which gave the inhibitor its name. The mitotic spindle is now a well-known target of chemotherapy, and inhibitors of the mitotic kinesins KIF11, KIF10 and KIF1C are being studied for this purpose.28 30 The redundancy of some kinesins allows them to escape pharmacological inhibition. For example, in the absence of KIF10, KIF15 is able to replace all of its essential functions in spindle assembly. Cilia-related KIF7, KIF13B and KIF27 are involved in SHh signalling and may be a future target in cancer research.28
Some kinesins confer susceptibility to a range of multifactorial, metabolic and neurodegenerative conditions. KIF13B contributes to the enhancement of endocytosis of low-density lipoprotein (LDL) receptor-related protein 1 that is involved in the recognition and internalisation of LDL and factor VIII. Kif13b-knockout mice have hypercholesterolaemia and higher factor VIII serum levels.5 KIF12 is implicated in the pathogenesis of type 2 diabetes, protecting pancreatic β cells from the oxidative stress caused by nutritional excess.5 Variants in KIF1B or KIF21B confer susceptibility to multiple sclerosis (OMIM %612596, #126200).31 32 KIF5A was associated with Amyotrophic lateral sclerosis (OMIM #617921).33 KIF3 complex and KIF17 were recently uncovered to be involved in schizophrenia.34 35 Further studies, however, are needed to clarify the precise role of KIFs in neurodegenerative processes and psychiatric conditions.
KIF14 -related birth defects: lessons learnt
Advances in next-generation sequencing technologies have revolutionised our understanding of Mendelian disorders, including birth defect phenotypes, by sequencing the coding genome (exome) or entire genome at an unprecedented resolution in a comparably short time span. The technology has been extensively used for gene identification approaches in research for many years, enabling unparalleled genotype–phenotype correlations and the definition of novel pathways of related genes and disorders at an accelerated pace, traditionally focusing on postnatal disorders. Filges and Friedman36 postulated that a number of novel disease genes causing birth defects could be identifiable through the investigation of lethal foetal phenotypes since they would represent the extreme end of allelic milder and viable postnatal phenotypes with less specific or recognisable anomaly patterns. Based on embryonically or perinatally lethal mouse models (www.informatics.jax.org and www.dmdd.org.uk), it is estimated that knockout variants in about 30% of human protein coding genes may present with a phenotype of early lethality. The identification of KIF14 loss of function variants in fetuses with a lethal multiple congenital anomaly syndrome and the subsequent description of the allelic postnatal viable phenotype and further functional characterisation of KIF14 in developmental processes are recent examples of how to study those embryonic lethal phenotypes in order to understand the role of genes for which little to nothing is known.
Filges et al identified autosomal recessive compound heterozygous loss of function variants in KIF14 using family-based exome sequencing in a recurrent severe lethal phenotype (OMIM #616258). It was the first human phenotype reported due to variants in the human KIF14 gene (figure 3).37 The two affected siblings presented with intrauterine growth retardation (IUGR), oligohydramnios, severe microcephaly, renal cystic dysplasia or agenesis, genital tract malformations (uterine hypoplasia and vaginal atresia), as well as cerebral and cerebellar hypoplasias with partial or total agenesis of the vermis, arhinencephaly, agenesis of occipital lobes/corpus callosum at second trimester ultrasound scan. Cross-species comparison to the laggard spontaneous mice mutant, characterised by homozygous variants of the Kif14 gene,38 confirmed a phenotypical overlap. An increased number of binucleated cells in the tissue histology of the two fetuses were in concordance with the key role of KIF14 during mitosis participating in chromosomes’ congression and alignment, as well as in cytokinesis39 and the observation of binucleated cells as a consequence of failed cytokinesis in mammalian KIF14 knockdown cells. During cytokinesis, PRC1 localises KIF14 at the central spindle and midbody, which in turn recruits citron rho-interacting kinase (CIT) to the midbody. CIT, in turn, acts as a negative regulator of KIF14 activity. Knockdown of KIF14 in mammalian cells results in impaired localisation of CIT during mitosis.40

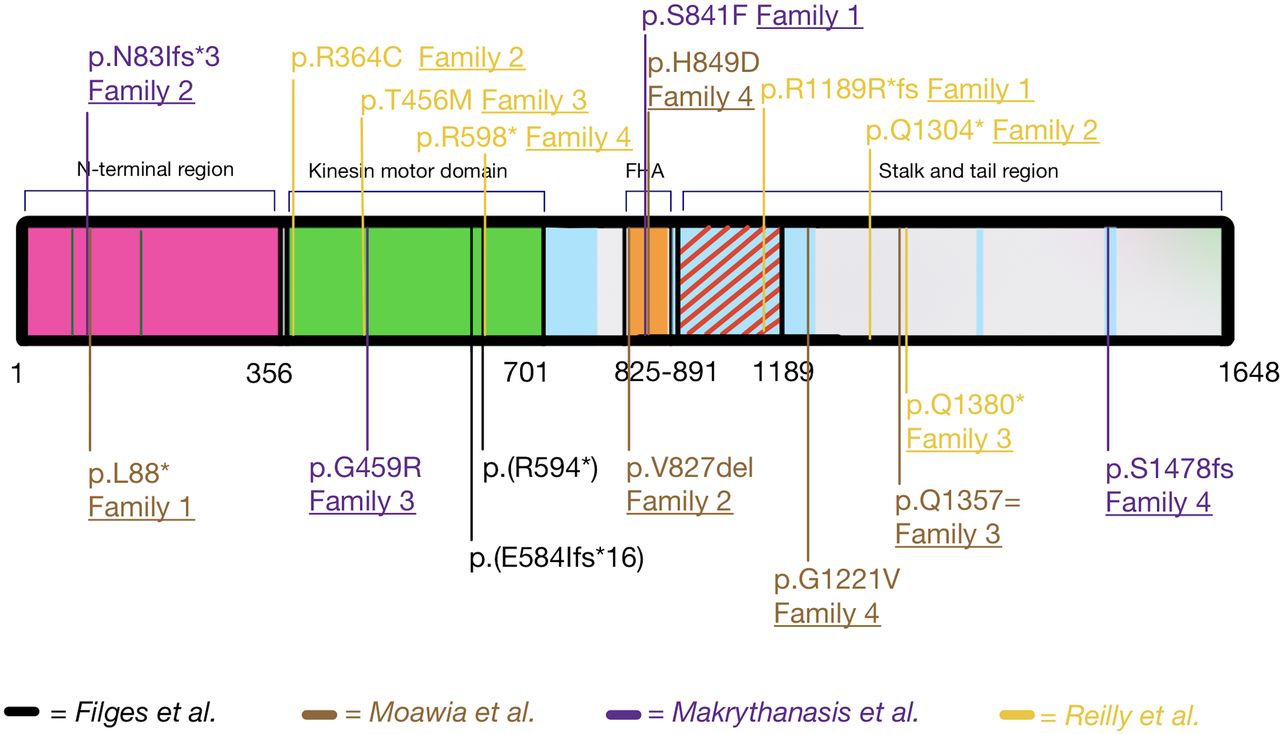
Structure of KIF14 and summary of all published KIF14 variants affecting function.10 37 41 42 The N-terminal region (aa 1–356) is important for its interactions with PRC1 and the protein’s localisation at the central spindle and midbody; the kinesin motor domain (aa 358–701) is responsible for the microtubule-dependent ATPase activity; the FHA domain (aa 825–891); stalk and tail region (aa 891–1648) are necessary for the interaction with the protein CRIK (aa 901–1189, red diagonal lines). There are four additional coiled-coil domains (light blue-coloured areas).61 FHA, forkhead associated. aa, amino acid.
Filges et al pointed out that KIF14 should be considered a candidate gene for viable postnatal phenotypes, including isolated microcephaly.34 Additional individuals with autosomal recessive variants in KIF14 and isolated primary microcephaly were then described9 41 42 (table 2).
Impaired cytokinesis, increased apoptosis and reduced cell motility were confirmed in cells from the described patients, pointing to a new cellular pathway in the pathogenesis of microcephaly.43 Apart from one case with small kidneys with increased echogenicity, none of these 18 patients had associated kidney anomalies. However, a targeted exome sequencing study in 204 unrelated patients with congenital anomalies of the kidney and urinary tract (CAKUT) reported two more cases of renal anomalies, bilateral hypoplasia or agenesis, caused by KIF14 variants.44 Further nine cases had an associated renal phenotype, which ranged from bilateral renal agenesis to cystic or non-cystic renal hypodysplasia.42 Table 2 and figure 3 summarise KIF14 variants and the associated phenotypes. Loss of function variants more likely lead to multiple congenital anomalies, while hypomorphic variants result in a milder phenotype without renal involvement, although phenotype–genotype correlations remain preliminary for the time being.
The phenotypical spectrum ranging from isolated primary microcephaly to congenital anomalies reminiscent of ciliopathy phenotypes suggested a complex role for KIF14 in developmental processes and raised a number of questions about the relationship between its established role in cell division and its possible function in ciliary pathways. Functional studies of absent KIF14 protein in the development of human foetal tissues and mutant zebrafish provided evidence for similarities and differences between mitotic events occurring during proliferation in the development of both brain and kidney.42 The observation that KIF14-stained midbodies accumulate within the lumen of the branch tips of ureteric buds in human foetal kidneys provided a key clue to better understand the mechanism through which the loss of KIF14 affects both brain and kidney development in humans. It was previously demonstrated that the secretion and accumulation of midbody remnants in the cerebrospinal fluid in mice during the early stages of brain development correspond to the amplification of neural progenitors.45 Kif14 mutant zebrafish phenotypes supported the hypothesis of a potential role for KIF14 in cilia. In vitro and in vivo analyses suggested that loss of kif14 causes ciliary anomalies through an accumulation of mitotic cells in ciliated tissues but failed to establish a direct functional link.21 42 Further mechanisms remain to be elucidated. Overexpression of KIF14 in various types of tumours was suggested to be a possible prognostic marker and a potential target for therapeutic purposes.46
Kinesinopathies in birth defect phenotypes: recurrent themes
In the last few years, an increasing number of variants in KIF genes were described to cause isolated as well as multiple congenital anomalies. There is a huge variability of phenotypes caused by variants even within the same gene. However, we can identify recurrent clinical signs that should alert the clinician to suspect a KIF gene-related disorder and the molecular geneticist to include KIF genes in multigene-panel and genome-wide sequencing approaches. This will become particularly relevant in prenatal and perinatal medicine, which focuses on the detection of structural anomalies in the fetus and the newborn by using ultrasound and MRI or autopsy when the outcome is lethal. We have summarised the predominant and recurrent structural anomalies in kinesinopathies reported so far that would likely become apparent during the foetal period in table 3 and the syndromic disorders in table 1.
KIF gene-related structural congenital anomalies recurrently described in prenatal phenotypes
Supplemental material
Consistent with the kinesins’ role in the development of the central nervous system (CNS), brain anomalies of various degrees are a frequent clinical sign, particularly microcephaly, but include lissencephaly, polymicrogyria, thinned or agenesis of the corpus callosum, arhinencephaly, cerebral hypoplasia or atrophy, cerebellar hypoplasia or atrophy, brainstem hypoplasia and a molar tooth sign on brain imaging.12 22 37 44 47–51
Primary microcephaly can be detected prenatally or at birth12 22 47 48 50 51 and can present as an isolated or syndromic condition as, for example, caused by variants in KIF14 9 or in KIF11 (microcephaly with or without chorioretinopathy, lymphoedema or mental retardation; OMIM #152950).48
KIF7 variants were related to macrocephaly in the presence of congenital hydrocephalus (hydrolethalus syndrome LS2, OMIM # 614120). Isolated hydrocephalus was reported for KIF4A in a single case.11
Foetal akinesia and arthrogryposis (KIF5C 12, KIF14 34 and KIF26B 50) are likely secondary to the neurological compromise of the fetus but can also appear as an early sign of abnormal CNS development, which should prompt specialist CNS sonographic and MRI evaluation of the fetus.
Further anomalies of the limbs include camptodactyly (KIF26B 50), clubfoot (KIF1A 51), rocker-bottom feet (KIF26B 50) and congenital lymphoedema of the limbs (dorsa of feet, lower extremities and, rarely, hands) in cases with KIF11 gene mutations.48 In particular, KIF7 gene variants have been related to various anomalies of the hands (tapered fingers, fifth finger clinodactyly, brachydactyly, preaxial or postaxial polydactyly, bifid terminal phalanges of the thumbs, spindle-shaped fingers, clinodactyly and soft tissue webbing) and feet (toe syndactyly, preaxial or postaxial polydactyly, and duplicated halluces).22
CAKUT and genital anomalies are reported in various kinesinopathies including renal agenesis or hypoplasia (KIF14 37 and KIF12 52), ureteral hypoplasia (KIF14 37), congenital megabladder (KIF12 52) and vesicoureteral reflux (KIF12 52), uterine hypoplasia and vaginal atresia (KIF14 37) and hypospadias and chordae (KIF16B 49).
IUGR is recurrently detected (KIF5C 12, KIF14 37, KIF10 53, KIF15 54 and KIF2A 12) and is particularly relevant when occurring simultaneously with one of the other recurrent clinical signs, indicating a potential syndromic KIF-related disorder. Oligohydramnios or polyhydramnios is most likely secondary to a primary organ anomaly.
There are a few kinesinopathy syndromes that have been specifically reported to be lethal, such as the ciliary phenotype (OMIM #616258), caused by variants in KIF14 34, and hydrolethalus syndrome (OMIM #614120), caused by variants in KIF7.22 However, lethality is usually closely related to the specific major anomalies, and it can be hypothesised that such a lethal phenotype will exist for all KIF gene-related disorders.
Developmental delay, intellectual disability, seizures, and sensory and motor disturbances of the peripheral nervous system, as well as eye anomalies, such as microphthalmy, optic nerve pallor, fibrosis of extraocular muscles and chorioretinopathy, will escape detection in the foetal period but are reported in postnatal patients.
Kinesin pathways in birth defects
Functional studies of kinesins in birth defects are still sparse, and little is known about their networks and pathways. In order to improve our understanding, we used the Ingenuity Pathway Analysis (IPA Qiagen, Redwood City, California, USA) to visualise and analyse the connections between the 13 kinesin motor proteins associated with structural congenital anomalies (KIF5C, KIF1A, KIF1BP, KIF14, KIF16B, KIF7, KIF4A, KIF11, KIF10, KIF26B, KIF12, KIF15 and KIF2A) and in up to 10 of each of their most significant downstream proteins. The connections are defined as protein–protein interactions, activation, regulation of binding, expression, localisation, phosphorylation, protein–RNA interactions, molecular cleavage, ubiquitination, protein–DNA interactions, inhibition, translocation and transcription. Figure 3 displays the results. We used the software Gephy55 to look for all possible interactions between all proteins of the network and also used the IPA data to retrieve the canonical pathways involved. Figure 4 and online supplementary material, table 4, summarise the results. KIF7, KIF14 and KIF12 are located within the same network, and because of multiple connections between themselves and their downstream proteins, it is not surprising that they are all involved in kidney anomalies. IPA data are based on current publications and are therefore subject to bias because proteins that are most interconnected are also most probably those that have been more extensively studied. However, we consider the KIF genes coding for proteins seeming less important within the network to be strong candidates for future studies of human developmental disorders.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
IPA of the 13 kinesins known to be involved in birth defects with respect to their position in the cell. Proteins displayed on the right side of the figure, below the tag ‘other’, are those for which no subcellular location is known. Birth defect-related kinesins and their connection with each other are highlighted in green. Light blue-coloured downstream proteins are those which are known to cause birth defects when altered. Yellow-coloured proteins are those involved in neurological disorders overlapping with the clinical features of kinesinopathies. The legend of the biological function associated with every molecule is displayed on the right. Path Designer by IPA was used for the figure design. IPA, Ingenuity Pathway Analysis.
Closing remarks and future perspectives
Novel KIF genes are increasingly identified, and there is a growing body of literature demonstrating the impact of kinesin dysfunction in human disease. We propose to introduce the term kinesinopathies for conditions caused by variants in KIF genes, since recurrent and common functional and phenotypical themes can be observed. In analogy to ciliopathies56 and rasopathies,57 the delineation of the clinical, genetic and functional hallmarks of kinesinopathies will be important to better recognise these conditions, to understand the pathomechanisms and to ultimately improve the clinical management of the patients. Previously, the unified view of the phenotype characteristics of ciliary dysfunction allowed a tremendous increase in awareness, both in clinic and research, and the further identification of yet unrecognised ciliary disorders and the genes and proteins involved in their pathogenesis.56
Remarkable progress was achieved in assigning function to kinesins through their study in isolated and multiple congenital anomaly phenotypes. They are one large superfamily of molecular motors out of three (kinesins, dyneins and myosins), which is of key importance in several fundamental cellular processes using microtubules as rails for directional anterograde intracellular transport, including its regulation and modulating signal transduction.5 Kinesin motors are most important for the movement of chromosomes along the spindles during chromosome segregation, regulation of spindle formation, cell division and cytokinesis. These essential and broad cellular functions are critical for many physiological processes such as neuronal function and survival, some ciliary functions and ciliogenesis, determination of the left/right asymmetry of our body and regulation of organogenesis, thus explaining the impact and emerging recognition of kinesins in embryonic and foetal development. Defects can result in neuropathies, higher brain functions and structural brain anomalies. Multiple congenital anomalies, including the kidney and urinary tract and limb anomalies, are repeatedly reported. Microcephaly, which is usually not associated with genes implicated in specific ciliary mechanisms, and CNS anomalies are the most recurrent clinical signs in both the prenatal and postnatal phenotypes described so far. The discovery of the implication of KIF14 in microcephaly further suggested a possible novel role of other microcephaly proteins in cytokinesis. A number of syndromic kinesinopathies present, however, with phenotype patterns reminiscent of ciliopathies. So far, however, a direct functional impact was confirmed in only a few and could not be demonstrated, for example, for KIF14, despite an overlapping clinical pattern. In turn, ciliopathies are a clinically and genetically heterogeneous group of conditions themselves. Studying tissue and cell type-specific function and expression may help to further define the specific defects related to the individual aberrant kinesin.
The pleiotropic nature of human kinesinopathies, however, is just emerging, but their study promises to provide important insights into human developmental pathways. Seemingly unrelated clinical entities are highlighting a common theme. In a relatively short time span, monogenic KIF-related disorders were identified to present with often severe and lethal antenatal anomalies, with multiple or isolated congenital anomalies, neurodevelopmental and neurological disorders, or an increased susceptibility to multifactorial conditions. We focused on the emerging role of kinesins in structural congenital anomalies because, as illustrated for the KIF14 gene, great potential to decipher allelic viable phenotypes and developmental pathways lies in the study of these human knockout phenotypes at the severe end of the phenotypical spectrum. Knockout variants in about 30% of human protein coding genes in our genome may present with a phenotype of early lethality, and KIF genes seem to play an important role in such fundamental processes of human development. Identifying and characterising the variants, genes and phenotypes will extend our knowledge on early human development and pathomechanisms, and will ultimately also improve the clinical utility of genome-wide sequencing approaches for prenatal and postnatal application by our increased ability to interpret loss of function and hypomorphic variants alike. Furthermore, kinesins were extensively studied in cancer research and therapeutic strategies targeting their specific functions, such as the example of monastrol and other inhibitors of the mitotic kinesins may be adopted in the future. There are likely many more kinesinopathies to be unravelled in the field of birth defects because of their pivotal role in cellular logistics, but their recognition in clinics and research will depend on our ability to identify and characterise the common clinical, molecular and functional themes of these disorders and to use them to improve our understanding of their disease mechanisms.
References
Footnotes
Contributors SK and IF have both developed the concept and written the manuscript.
Funding The Swiss National Science Foundation (project grant number 320030_160200 awarded to IF) supported this work.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.