Article Text

Abstract
Background Breast cancer (BC) is the most common malignancy in women and has a major heritable component. The risks associated with most rare susceptibility variants are not well estimated. To better characterise the contribution of variants in ATM, CHEK2, PALB2 and XRCC2, we sequenced their coding regions in 13 087 BC cases and 5488 controls from East Anglia, UK.
Methods Gene coding regions were enriched via PCR, sequenced, variant called and filtered for quality. ORs for BC risk were estimated separately for carriers of truncating variants and of rare missense variants, which were further subdivided by functional domain and pathogenicity as predicted by four in silico algorithms.
Results Truncating variants in PALB2 (OR=4.69, 95% CI 2.27 to 9.68), ATM (OR=3.26; 95% CI 1.82 to 6.46) and CHEK2 (OR=3.11; 95% CI 2.15 to 4.69), but not XRCC2 (OR=0.94; 95% CI 0.26 to 4.19) were associated with increased BC risk. Truncating variants in ATM and CHEK2 were more strongly associated with risk of oestrogen receptor (ER)-positive than ER-negative disease, while those in PALB2 were associated with similar risks for both subtypes. There was also some evidence that missense variants in ATM, CHEK2 and PALB2 may contribute to BC risk, but larger studies are necessary to quantify the magnitude of this effect.
Conclusions Truncating variants in PALB2 are associated with a higher risk of BC than those in ATM or CHEK2. A substantial risk of BC due to truncating XRCC2 variants can be excluded.
- Cancer: breast
- Genetic Epidemiology
- Evidence Based Practice
- Geneticscreening/counselling
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/
Statistics from Altmetric.com
Introduction
Breast cancer (BC) is the most common malignancy and the second leading cause of cancer deaths in women worldwide.1 Both twin and population-based family studies indicate a substantial heritable component to BC risk.2 To date, approximately half of familial risk of BC has been explained by a combination of common variants with small effect sizes,3 together with rarer, protein coding variants that confer higher risks.4 Truncating variants in some genes, including BRCA1 and BRCA2, are associated with very high absolute risks, while deleterious variants in other genes, including ATM, CHEK2 and PALB2, have been reported to confer more moderate risks.4 However, since susceptibility alleles in these genes are rare, the risks associated with these have not yet been well estimated. Variants in several other genes, including XRCC2, have been suggested to contribute to risk, but evidence is more equivocal.4
ATM, CHEK2, PALB2 and XRCC2 play important roles in DNA repair. The ATM gene encodes a protein kinase that recognises double stranded DNA breaks and initiates multiple aspects of the damage response cascade. A recent meta-analysis estimated that truncating ATM variants were associated with a relative risk of 2.8,4 and several studies have reported that certain subgroups of missense variants also contribute to BC risk.5–9 CHEK2 encodes a checkpoint kinase that interacts with cell cycle regulators and DNA repair proteins. The most common protein-truncating genetic variant in Western European populations is the frame shift c.1100delC (p.Thr367MetfsTer15, rs555607708); a recent analysis by the Breast Cancer Association Consortium (BCAC) estimated a relative risk of 2.26 for this variant.10 Truncations of PALB2, the partner and localiser of BRCA2, have been associated with a fivefold relative risk,4 though studies in different populations have produced divergent estimates.11–14 XRCC2 encodes a protein in the Rad51 family that participates in homologous recombination repair of double-stranded DNA breaks. Protein-truncating mutations in this gene are uncommon, but an association with BC risk has been suggested for rare missense variants.15
These four genes are now included in most multigene BC risk sequencing panels,4 and it is therefore critical for genetic counselling to have accurate estimates of the BC risk associated with variants in these genes. To provide such estimates, we undertook a large, population-based study in which we sequenced the protein coding exons and intron-exon boundaries of ATM, CHEK2, PALB2 and XRCC2 in 13 087 BC cases and 5488 controls. We evaluated sets of rare variants, defined by predicted functional effect, and more common individual variants (population frequency >0.1%), for association with BC risk.
Materials and methods
Study population
Cases were drawn from SEARCH, a population-based study of BC in the region of East Anglia (UK) covered by the Eastern Cancer Registration and Information Centre (ECRIC).16 The study enrolled subjects diagnosed before age 55 years with invasive BC from 1991 onwards and who were still alive at the start of the study in 1996 (prevalent cases, n=1087; median age=48 years), together with all patients diagnosed before age 70 years between 1996 and the present (n=12 000). Data on oestrogen-receptor and progesterone-receptor status and bilaterality were obtained through ECRIC and abstraction of medical records. Controls were drawn from three sources: (1) general practices participating in SEARCH who were frequency matched by age to the cases; (2) the European Prospective Investigation of Cancer (EPIC)-Norfolk study, a population-based cohort study of diet and health in Norfolk, East Anglia;17 and (3) women undergoing breast screening as part of the National Health Service Breast Screening Programme in screening centres in Cambridgeshire, who participated in the Sisters in Breast Screening study.18 Sequence analysis was conducted on samples from 13 824 BC cases and 5952 controls (pooled from the three studies above) of which 13 087 cases and 5488 controls passed all QC filters (see online supplementary figure s1 and supplementary methods) and were used in the analysis. Ethics approval was provided by the Cambridgeshire Research Ethics Committee, and written consent was obtained at the time of sample collection.
Amplicon design, enrichment, sequencing and variant calling
The Fluidigm Access Array 48.48 system was used for library preparation (see online supplementary methods). We designed 211 amplicons (see online supplementary table S1) to cover 98.1% of the bases within Consensus Coding Sequence (CCDS) exons of the four genes (see online supplementary table S2). Each library of 211 amplicons for 1536 samples was sequenced in 100-base paired-end mode on a single lane of an Illumina Hi-Seq2000. Raw sequence data were demultiplexed using the Illumina CASAVA 1.8 pipeline and aligned to the hg19 human reference sequence with BWA-MEM V.0.7.19 GATK UnifiedGenotyper was used to perform SNP and indel discovery and variant calling across all samples simultaneously (see online supplementary methods).20–22 After filtering samples and variants with >5% missing calls, 5488/5952 controls (92%) and 13 087/13 824 cases (95%) were retained for further analysis. Final variant calls were reproducible within this study and concordant with orthogonal methods (see online supplementary table s3 and supplementary methods).
Functional prediction and variant frequency classification
The Ensembl Variant Effect Predictor23 was used to assign the canonical transcript-level and protein-level consequence for each genetic variant. Frameshift, stop/gain and canonical splice variants were grouped as protein truncating. Missense variants were further annotated with effect predictions from CADD,24 PolyPhen2,25 SIFT,26 and AlignGVGD.27 The consequences of the putative splice site variant CHEK2 c.320–5T>A were evaluated using the in silico prediction tools SpliceSiteFinder-like,28 MaxEntScan,29 NNSPLICE,30 GeneSplicer31 and Human Splicing Finder.32
Variants detected in these four genes were annotated with allele frequencies observed in populations catalogued in the Exome Aggregation Consortium (ExAC) variation database (http://www.exac.broadinstitute.org).33 All coding variants with a carrier frequency >0.1% in the 33 370 non-Finnish, European ExAC subjects were classified as common, while all others (including those not reported by ExAC) were classified as rare. The combined East Anglian case and control frequency of <0.1% was used to define rare non-coding variants.
Statistical analysis
For each variant with a carrier frequency >0.1% in ExAC European subjects (coding variants) or study cases and controls combined (non-coding variants), per-allele ORs and 95% CIs were computed, and Cochran-Armitage trend tests were carried out using the ‘prop.trend.test’ module of R.34
Since most variants were too rare to derive individual risk estimates, variants were grouped into classes (truncating, missense, synonymous and non-canonical splice) based on their predicted effect, and estimates were derived for carriers of any variant in each class. Rare missense variants were further subdivided based on domain, functional prediction scores and protein position. ORs were estimated by unconditional logistic regression, using the glm package in R. Profile likelihood-based confidence limits were derived using the confint routine. ORs were estimated using both the study controls (n=5488) and non-Finnish European ExAC subjects (n=33 370).
For truncating variants, separate analyses were conducted for oestrogen receptor (ER)-negative and ER-positive BC. Differences in ORs by subtype were assessed using case-only analyses.
Results
Spectrum of variation
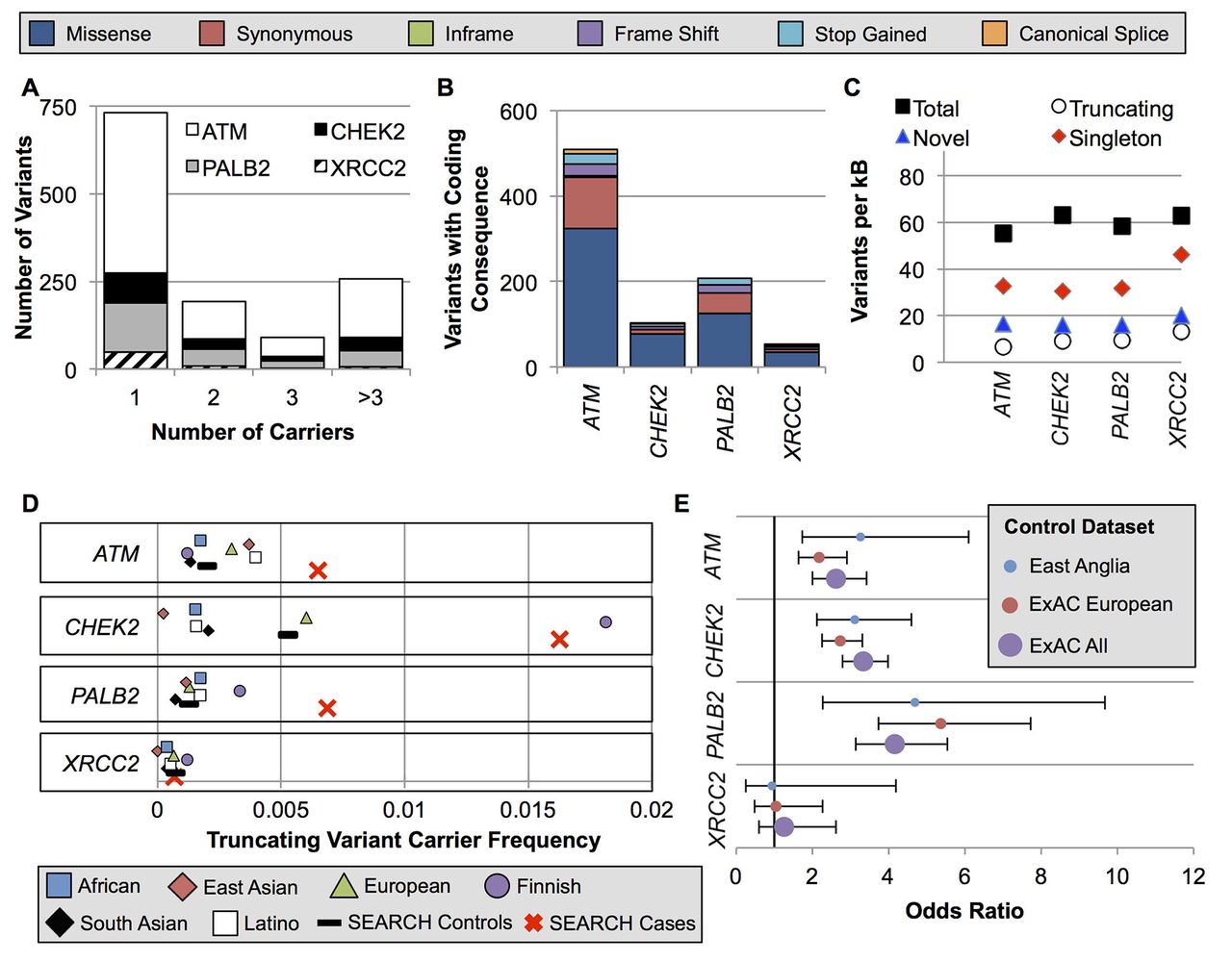
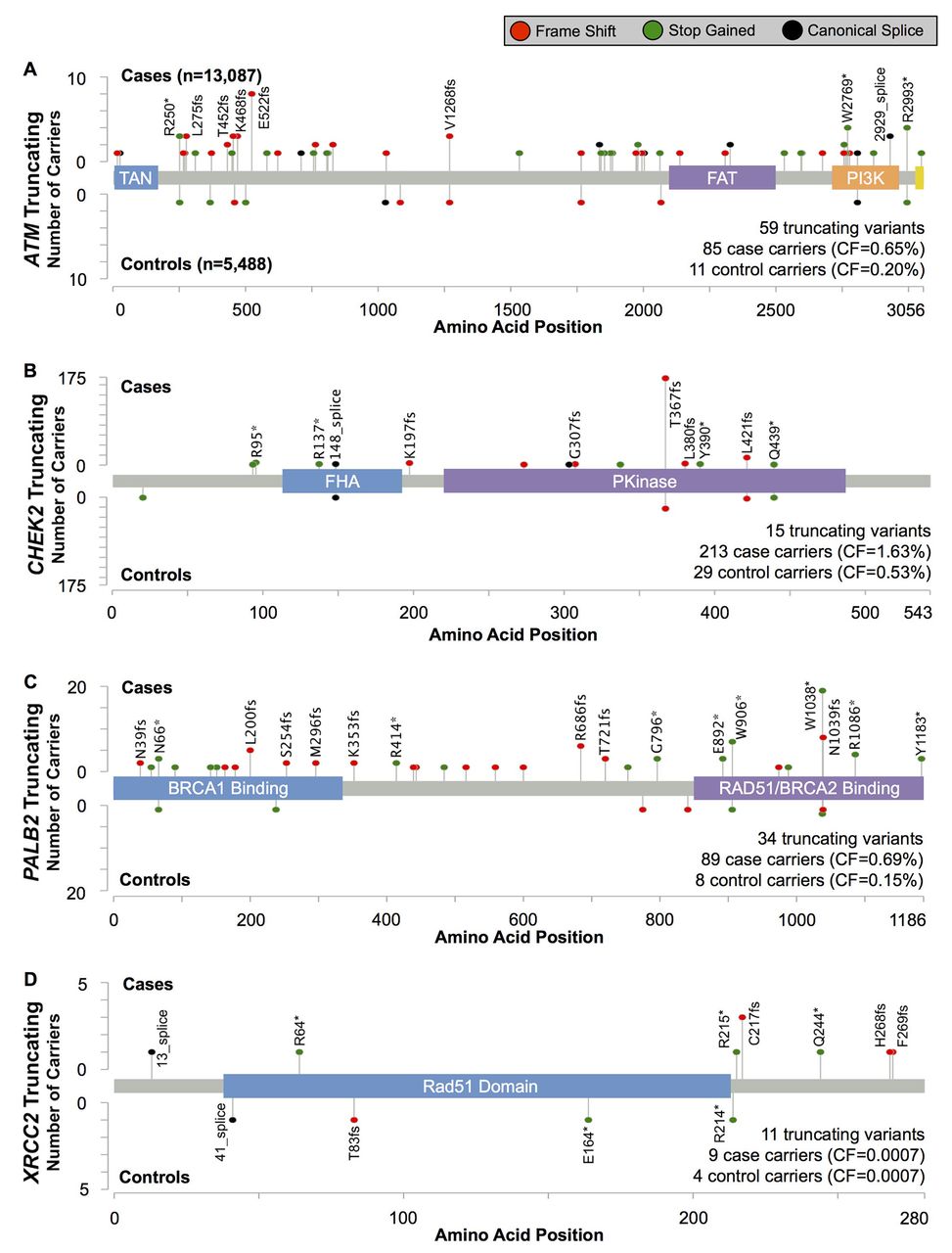
We identified 1273 variants in the four genes: 785 in ATM, 165 in CHEK2, 255 in PALB2 and 68 in XRCC2 (online supplementary tables S4 and S5). Among these, 72 were common variants (for coding variants: carrier frequency >0.1% in ExAC non-Finnish, European ancestry subjects; for non-coding variants: carrier frequency >0.1% in cases and controls combined) (see online supplementary table S4). However, the majority of variants (731/1273, 57.4%) were identified in a single subject (figure 1A). Most variants encoded a missense substitution (figure 1B), and 29.1% of the coding variants identified in this study had not been previously reported in the dbSNP, ExAC or COSMIC databases (figure 1C). All four genes had similar variant rates after adjusting for gene length, with approximately 60 variants per kilobase of coding sequence (figure 1C).
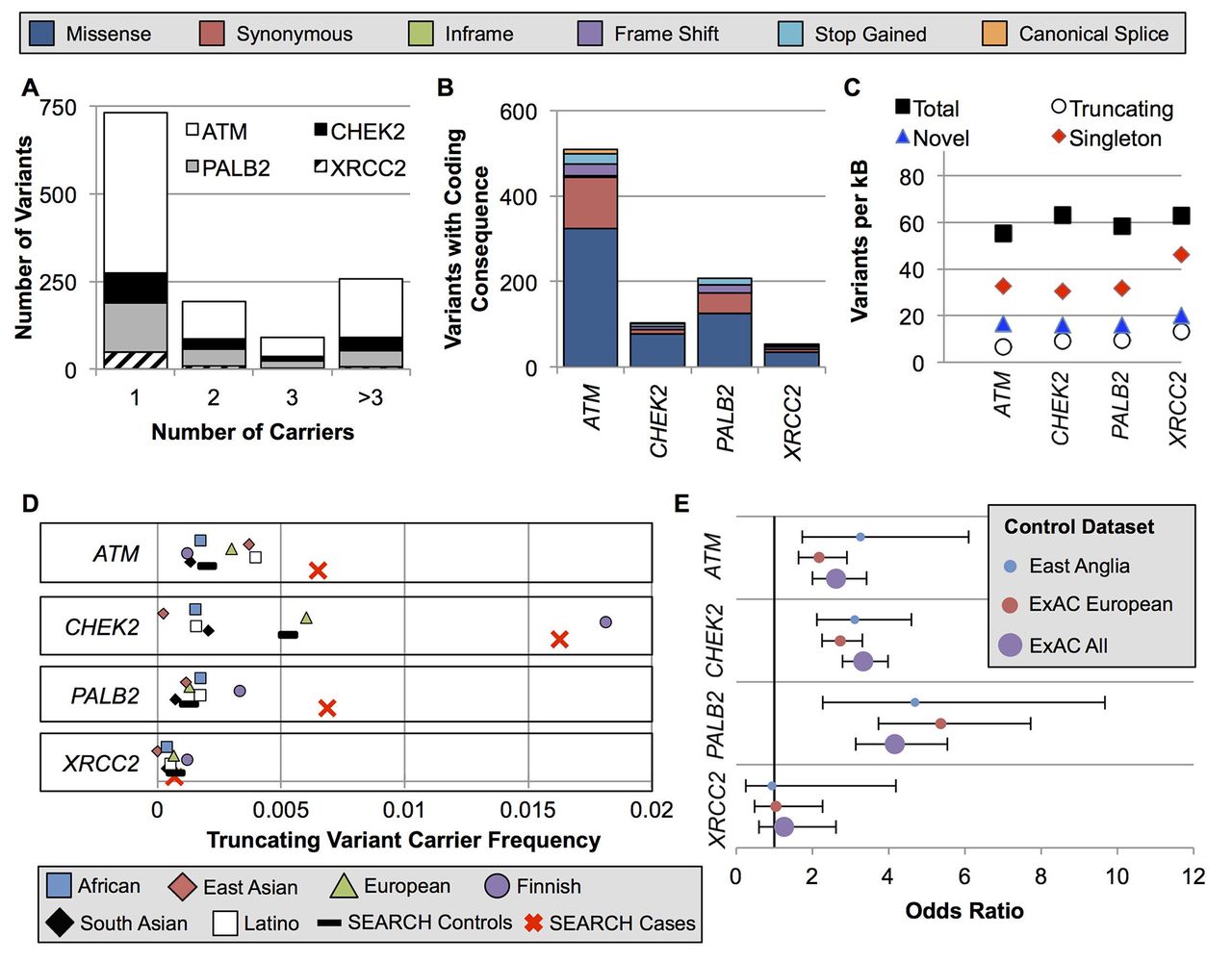
Spectrum of ATM, CHEK2, PALB2 and XRCC2 variation in ExAC populations and East Anglian cases and controls. (A) Nearly all variants were very rare or private. (B) The absolute number of variants per gene was highly divergent due to the difference in gene size. (C) However, when corrected for gene size, the genes had similar per-kB rates of total, truncating, singleton and novel variants not previously reported in the dbSNP, ExAC or COSMIC. (D) The carrier frequency for truncating variants in the study controls was near the mean of the ExAC populations. Notably, the ExAC Finnish population was an outlier for CHEK2 and PALB2 due to well-studied founder effects. (E) OR point estimates were similar using the study controls versus ExAC populations as controls. ExAC, Exome Aggregation Consortium.
BC risks associated with ATM, CHEK2 and PALB2 truncating variants
Of the four genes, truncating variants in PALB2 were associated with the highest BC risk, with an estimated OR=4.69 (95% CI 2.27 to 9.68, p=6.9×10−6)(table 1 and figure 2). Most truncating variants were rare, with 19/35 (54%) observed as singletons. Three variants, all in the final 280 codons of the gene, were more common: PALB2 c.2718G>A (p.Trp906Ter, rs180177122; seven cases, one control), c.3113G>A (p.Trp1038Ter, rs180177132; 20 cases, one control) and c.3116delA (p.Asn1039IlefsTer2, rs180177133; eight cases, one control) (see online supplementary table 5). The combined OR for these three more common, C-terminal truncations was 3.57 (95% CI 1.27 to 10.1, p=0.016), somewhat, but not significantly, lower than the point estimate for all other truncating variants in the gene (OR=5.79; 95% CI 2.10 to 16.0, p=2.2×10−4; Pdiff=0.78).
Overall BC risk estimates for truncating mutations in each gene. Variant category tallies (online supplementary table S4) and carrier counts (online supplementary table S5) are available in the supplementary materials
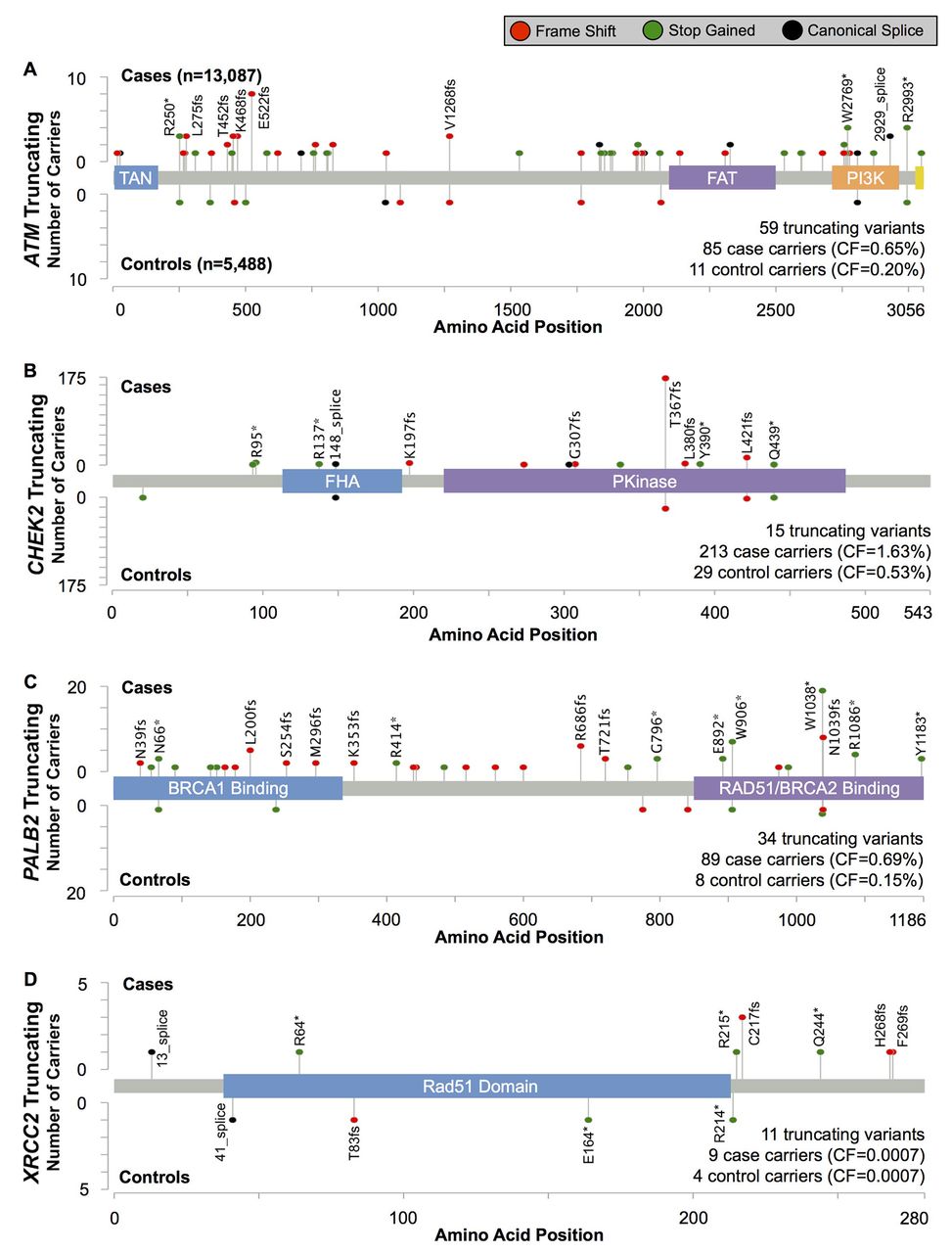
Position and frequency of protein-truncating variants in (A) ATM, (B) CHEK2, (C) PALB2 and (D) XRCC2.
The risk estimate for CHEK2-truncating variants was OR=3.11 (95% CI 2.15 to 4.69, p=5.6×10−11; table 1 and figure 2). The most common truncating variant, CHEK2 c.1100delC (p.Thr367MetfsTer15, rs555607708) accounted for 81% (196/242) of truncating variant carriers (online supplementary table S5) and was associated with an OR=3.18 (95% CI 2.01 to 4.92, p=6.1×10−8). The risk estimate for the aggregate of the remaining 14 rare truncating variants was OR=2.83 (95% CI 1.20 to 6.69, p=0.020), consistent with the estimate for CHEK2 c.1100delC.
Protein-truncating variants in ATM were associated with an elevated risk of BC similar to CHEK2 (OR=3.26; 95% CI 1.82 to 6.46, p=2.1×10−5; table 1 and figure 2).
Only 13 carriers of truncating variants in XRCC2 were found in our study (table 1 and figure 2). There was no evidence for association with BC risk (OR=0.94; 95% CI 0.26 to 4.19, p=0.92).
For each of the four genes, the truncating variant carrier frequencies in the ExAC non-Finnish European population were similar to those in the study control group. Consequently, similar OR estimates were obtained when this dataset was substituted for the study controls (figure 1D–E and see online supplementary table S6).
BC associations for protein truncating variants disease subtype, age and family history
Truncating variants in CHEK2 were associated with a higher relative risk for ER-positive (OR=3.42; 95% CI 2.33 to 5.21; table 2), and lower, non-significant risk for ER-negative BC (OR=1.59; 95% CI 0.80 to 3.00; Pdiff=0.0032). A similar pattern was observed for progesterone receptor status, though the difference was not significant (Pdiff=0.18). Truncating variants in ATM were also associated with a higher risk for ER-positive disease (OR=3.42; 95% CI 2.33 to 5.21) than ER-negative disease (OR=1.59; 95% CI 0.80 to 3.00), though not significantly so (Pdiff=0.11). There was no evidence of a difference in the estimated OR by ER-status for PALB2 variants (ER-positive OR=4.32; 95% CI 2.07 to 10.5 vs ER-negative OR=5.58; 95% CI 2.19 to 15.2; Pdiff=0.55).
Associations by family history, synchronous or metachronous bilateral disease, age at diagnosis and hormone receptor subtype.
Truncating ATM variants were more common in BC cases with a family history of BC among first-degree relatives (OR=2.06; 95% CI 1.12 to 3.64, p=0.022; table 2). There was some evidence of an association with a positive family history for carriers of truncating CHEK2 variants (OR=1.51; 95% CI 0.97 to 2.28, p=0.070), but not for PALB2 truncation carriers (OR=0.74; 95% CI 0.60 to 1.54, p=0.44). Bilateral BC was more common than unilateral disease in women with CHEK2-truncating variants (OR=3.27; 95% CI 1.66 to 5.83, p=0.0014; table 2). There was some evidence of an association with bilaterality for PALB2 variant carriers (OR=2.85, 95% CI 0.86 to 6.91, p=0.080). No ATM truncations were found among bilateral BC cases.
The relative risk associated with CHEK2-truncating variants declined with increasing age, with estimated ORs of 3.98 for diagnosis before age 50, 3.37 between ages 50 and 59 and 2.12 after age 60 (Ptrend=1.2×10−5; table 2). For ATM and PALB2 variant carriers, there was no clear evidence for an OR trend by age (Ptrend=0.66 and 0.22, respectively).
Risk associations for subsets of missense variants
Thirty-eight missense variants had an ExAC carrier frequency >0.1% (25/299 in ATM, 4/77 in CHEK2, 7/125 in PALB2 and 2/34 in XRCC2; see online supplementary table S4), and two were significantly associated with BC risk: ATM c.7390T>C (p.Cys2464Arg, rs55801750, OR(Arg/Cys)=0.37; 95% CI 0.19 to 0.73, Ptrend=0.0028) and XRCC2 c.563G>A (p.Arg188His, rs3218536, OR(His/Arg)=0.90; 95% CI 0.83 to 0.97, Ptrend=0.0080) (see online supplementary table S7). However, neither of these associations was significant after adjusting for multiple testing. Of note, CHEK2 c.470T>C (p.Ile157Thr, rs17879961) was found in 0.13% of subjects (20 cases, four controls; see online supplementary table S5) in this study. The relative risk estimate (OR(Thr/Ile)=2.10; 95% CI 0.72 to 6.14, p=0.17), although non-significant, is compatible with the 1.4-fold increased risk previously reported for the same variant in Finnish and Eastern European populations.3 35
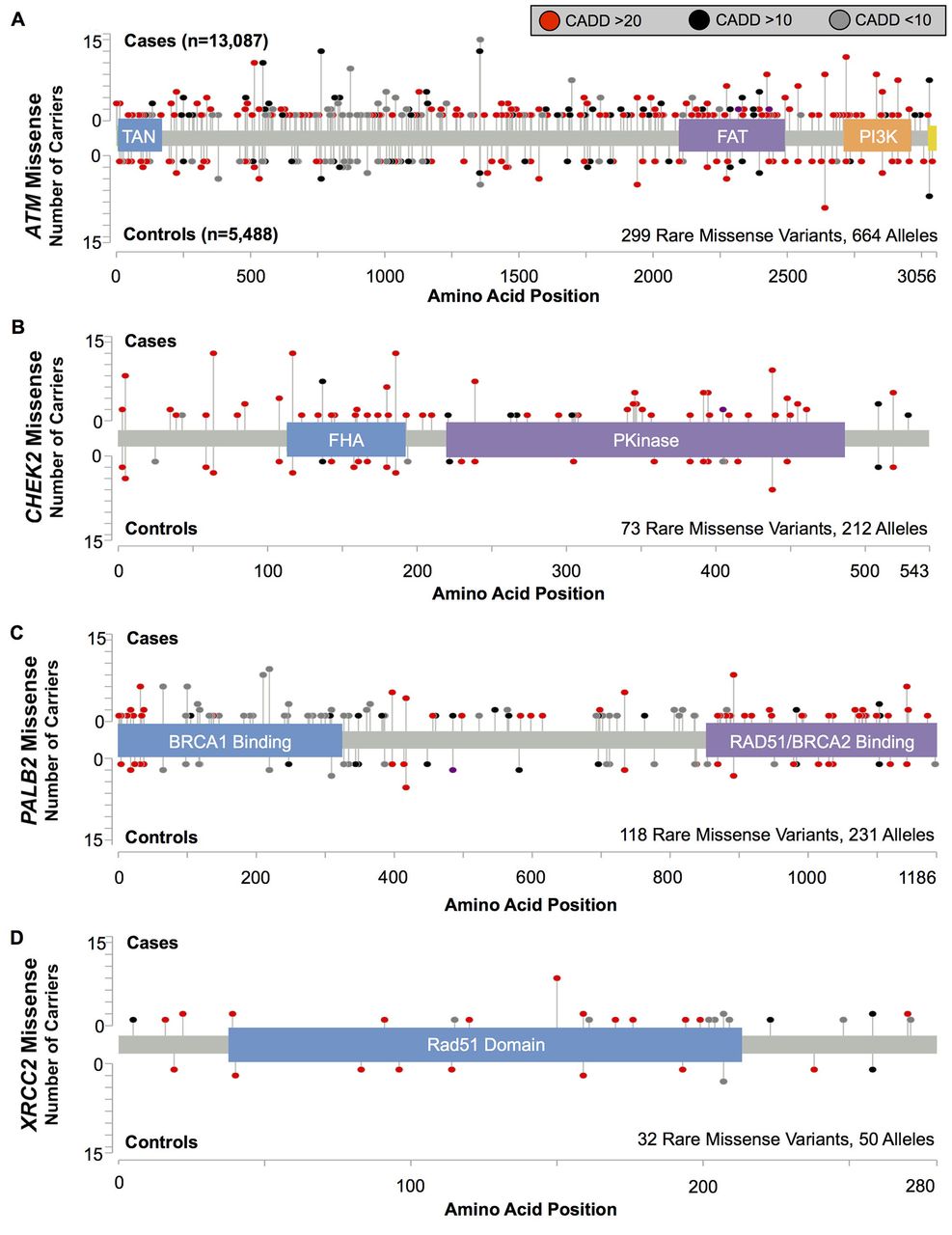
We tested for risks associated with the aggregate of all rare missense variants in each gene, irrespective of position or predicted deleteriousness (figure 3 and table 3). We found some evidence of increased BC risk associated with the combined rare missense substitutions in ATM (OR=1.18; 95% CI 0.99 to 1.40, p=0.073), CHEK2 (OR=1.36; 95% CI 0.99 to 1.87, p=0.066) and PALB2 (OR=1.28; 95% CI 0.95 to 1.73, p=0.12), but not in XRCC2. Considered together, rare missense variants in ATM, CHEK2 and PALB2 were associated with an estimated OR=1.24; 95% CI 1.08 to 1.43, p=0.0025 (table 3). Variants localised within protein domains (as defined by UniProt or Pfam) of these three genes had a somewhat higher risk estimate (OR=1.45; 95% CI 1.17 to 1.80; figure 3 and table 3) than those outside of the annotated domains (OR=1.09; 95% CI 0.92 to 1.31; Pdiff=0.060).
{kind=link}
{kind=link}
{kind=link}
Position and frequency for rare missense variants in (A) ATM (B) CHEK2 and (C) PALB2. ORs were calculated for all Pfam (ATM and CHEK2) and UniProt (PALB2) domains (table 2).
Rare missense variant associations with BC by domain
There was no evidence that risk was higher among variants predicted to be deleterious by CADD, PolyPhen2, SIFT or AlignGVGD, for any of the four genes, and no subset of variants stratified by these annotations was significantly associated with risk (see online supplementary table S8). Similarly, the risk estimate for the aggregate of rare variants in ATM, CHEK2 and PALB2 with deleterious functional predictions was not significantly higher than for predicted benign variants (three genes combined Pdiff=0.91, 0.74, 0.71 and 0.76 for CADD, PolyPhen, SIFT and AlignGVGD, respectively; see online supplementary table S8).
Previous analyses have indicated that ATM missense variants within the FRAP-ATM-TRRAP (FAT) and phosphatidylinositol 3-kinase (PI3K) domains were specifically associated with increased BC risk.5 6 We found evidence for increased risk for variants in both these domains (combined OR=1.71; 95% CI 1.12 to 2.61, p=0.015), but estimates did not differ significantly from those for the aggregate of all rare missense variants (Pdiff=0.31). Of note, c.7271T>G (p.Val2424Gly, rs28904921), which has been implicated in a milder Ataxia-Telangiectasia disease phenotype and has previously been associated with a substantial BC risk,5 7 8 occurred in eight cases and no controls in our study (figure 3 and see online supplementary table S5). After excluding this variant, the remaining rare missense substitutions in the FAT and PI3K domains in aggregate were still associated with BC risk (OR=1.59; 95% CI 1.04 to 2.43, p=0.040).
In PALB2, missense variants within the N-terminal BRCA1 binding domain were most strongly associated with risk (OR=1.76; 95% CI 1.03 to 2.98, p=0.047). This signal was driven by rare missense variants (n=29) between amino acids 70 and 300, and few of these were predicted by CADD, PolyPhen2, SIFT or AlignGVGD to have a deleterious effect on the protein (see online supplementary table S5).
Non-canonical splice variants and BC risk
We also examined associations for common variants in non-coding regions (see online supplementary table S7). Among these, only CHEK2 c.320–5T>A (rs121908700; OR=13.9; 95% CI 1.89 to 101, Ptrend=6.7×10−4) was significantly associated with risk after correction for multiple testing. This variant, in a non-canonical splice site, was predicted to reduce recognition of the normal splice acceptor site of exon 3 and introduce a new acceptor site three nucleotides upstream. At the protein level, this change would preserve the reading frame and cause the insertion of a valine residue. There was some suggestion of an association for the aggregate of other non-canonical splice variants in CHEK2, which were found in 12 cases and two controls (OR=2.52, 95% CI 0.56 to 11.3, p=0.26; online supplementary table S5).
Discussion
This study, the largest experiment to date to systematically sequence the coding and exon-flanking regions of these genes in a population-based series of BC cases and controls, provides additional confirmation that protein-truncating mutations in ATM, CHEK2 and PALB2 are associated with increased BC risks. For ATM and CHEK2, the relative risks were higher for ER-positive than ER-negative disease, but we observed no differential effect by ER-status for PALB2. In contrast, XRCC2-truncating variants were not significantly associated with risk, but a twofold increased risk could not be excluded because these variants were very rare (13/18 575 samples; upper 95% confidence limit 4.19). These findings underscore the fact that, despite the large size of the study, the data are too sparse to accurately estimate risks for very rare variant classes and less common BC subtypes (eg, triple negative disease).
The BC risk estimates for all three of the associated genes were similar to estimates from smaller case–control studies and studies based on family-based designs (in the combined analysis of previous studies reported by Easton et al 4: PALB2: meta analysis OR=5.3; 95% CI 3.0 to 9.4 vs table 1 OR=4.69; 95% CI 2.27 to 9.68; ATM: meta analysis OR=2.8; 95% CI 2.2 to 3.7 vs table 1 OR=3.26; 95% CI 1.82 to 6.46; and CHEK2: meta analysis OR=3.0; 95% CI 2.6 to 3.5 vs table 1 OR=3.11; 95% CI 2.15 to 4.69). Based on the estimated population frequencies and relative risks from this study, truncating variants in ATM, CHEK2 and PALB2 would explain approximately 4% of the twofold familial relative risk of BC and approximately 2% of all BC cases. While these estimates were derived from a study in the UK, the comparability of the combined frequency of truncating variants in our study with those from ExAC suggests that these estimates are likely to be broadly applicable to other European populations. Somewhat surprisingly, we observed no association between carrying a truncating PALB2 variant and a BC family history, but this may reflect lack of power: there were only 53 carriers for whom family history data were available.
The vast majority of the truncating variants in this study were very rare: 117/119 were found in <0.1% of samples. The most notable exception was CHEK2 c.1100delC, which was identified in approximately 1.1% of subjects and accounted for 81% of truncation carriers in this gene. Our risk estimate for this variant (OR=3.18; 95% CI to 2.01 to 4.92) was somewhat higher than two recent analyses (BCAC: OR=2.26; 95% CI 1.90 to 2.6910; Danish cohort: OR=2.08; 95% CI 1.51 to 2.85).36 These differences might be explained by, for example, differences in the age distribution of the study subjects. The risk estimate for aggregated non-c.1100delC truncating variants in CHEK2 was similar to that for c.1100delC, suggesting that results for this founder variant can reasonably be extrapolated to other truncating variants.
No individual missense variants showed evidence of association with BC risk at p<0.001, nor did we find strong evidence for the aggregate of rare missense variants in a single gene. There was, however, an association with BC risk for all rare, non-synonymous substitutions combined across ATM, CHEK2 and PALB2. This risk could be mediated by a small subset of variants conferring a high risk, or a larger subset of variants associated with a lower risk. We observed little evidence of association by predicted effect severity, but there was, however, some suggestion that rare missense variants within functional domains may contribute to BC risk.
Conclusions
This report, based on a large population-based study, provides relative risk estimates associated with truncating variants in ATM, CHEK2 and PALB2. Our results confirm that risk estimates for ATM and CHEK2 gene variants are similar and firmly within the twofold to fourfold range. PALB2 protein-truncating variants conferred a somewhat higher risk, supporting previous suggestions that specific management may be justified in PALB2 carriers.11 The absolute risks and age-specific penetrance in carriers will depend on additional influences, including common susceptibility variants, lifestyle risk factors and family history—considerations that can be built into more comprehensive risk prediction models.37 Clinically useful risk estimates for rarer disease subtypes and for missense variants will require studies that are substantially larger than the current experiment; these are becoming possible through large consortia and technological advances.
Acknowledgments
BD is a student in the NIH-Cambridge Scholars Program. BD and EAO were supported by the Intramural Research Program of the National Human Genome Research Institute. SEARCH is funded by a programme grant from Cancer Research UK (C490/A10124) and supported by the UK National Institute for Health Research Biomedical Research Centre at the University of Cambridge. Targeted sequencing in SEARCH was supported by Cancer Research UK grants C1287/A16563 to DFE and C8197/A16565 to AMD. We thank Maaike Vreeswijk for her help with the in silico predictions for the CHEK2 splice variant rs121908700.
References
Footnotes
Contributors BD designed the targeted sequencing panel, performed bioinformatics and statistical analyses, and drafted the manuscript. JA performed bioinformatics analysis. CL and AMD coordinated the targeted sequencing and AMD also assisted with drafting the manuscript. KAP and SA contributed Sanger validation of variant calls. MS, MKB and QW provided data management support. CB, DMC and JB prepared targeted sequencing libraries. RL provided samples and phenotypic data from EPIC. EAO assisted with drafting the manuscript. PDPP coordinated SEARCH. DFE conceived and designed the study, performed statistical analysis and assisted with drafting the manuscript. All authors read and approved the final manuscript.
Funding Cancer Research UK (C490/A10124, C1287/A16563, C8197/A16565), NIH, NHGRI
Competing interests No, there are no competing interests.
Patient consent Obtained.
Ethics approval Cambridgeshire Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Collaborators Maaike Vreeswijk.