Article Text

Abstract
Objectives: To describe the clinical findings and natural history in 22 carriers of an R460H mutation in the transforming growth factor β receptor 2 gene (TGFβR2) from a five-generation kindred ascertained by familial aortic dissection.
Methods: 13 of the confirmed carriers were interviewed and examined, and information about the remaining carrier was obtained from medical records. Clinical information about deceased individuals was obtained, when possible, from postmortem reports, death certificates and medical records.
Results: There have been eight sudden deaths; the cause of death was aortic dissection in all six cases in which a postmortem examination was performed. Three individuals had undergone aortic replacement surgery. Dissection had occurred throughout the aorta, and in one case in the absence of aortic root dilatation. Subarachnoid haemorrhage, due to a ruptured berry aneurysm, had occurred in two individuals. Four gene carriers and one deceased family member who were investigated had tortuous cerebral blood vessels. One had tortuous vertebral arteries, two had tortuous carotid arteries and one a tortuous abdominal aorta. Two individuals were found to have a brachiocephalic artery aneurysm and a subclavian artery aneurysm, respectively.
Conclusions: Despite the predisposition to aortic dilatation and dissection, individuals did not frequently manifest the skeletal features of Marfan syndrome, with the exception of joint hypermobility. No one individual had ocular lens dislocation. Striae and herniae were common. There was some overlap with Ehlers–Danlos syndrome type 4, OMIM 130050, with soft translucent skin, which is easily bruised. Other features were arthralgia, migraine and a tendency to fatigue easily, varicose veins and prominent skin striae. This family provides further evidence that mutations in TGFβR2 cause a distinct syndrome that needs to be distinguished from Marfan syndrome to direct investigation and management of patients and shows the natural history, spectrum of clinical features and variable penetrance of this newly recognised condition.
- FBN, fibrillin gene
- TAAD2, thoracic aortic aneurysms and dissections
- TGFβ, transforming growth factor β
- TGFBR2, transforming growth factor β receptor 2 gene
Statistics from Altmetric.com
- FBN, fibrillin gene
- TAAD2, thoracic aortic aneurysms and dissections
- TGFβ, transforming growth factor β
- TGFBR2, transforming growth factor β receptor 2 gene
In 1993, Boileau et al1 reported on a large French family with a Marfan-like disorder that did not map to fibrillin 1 gene (FBN1) or FBN2. A year later, this family was mapped to 3p24.2–25 and this was proposed as a second locus for Marfan syndrome2 and the condition was designated Marfan syndrome type 2 (MFS2), OMIM 154705. Interestingly, a family with familial thoracic aortic aneurysms and dissections (TAAD2) have also been shown to map within this region.3
The transforming growth factor β receptor 2 (TGFβR2) gene was implicated in the pathogenesis of Marfan syndrome when Mizuguchi et al4 reported on a Japanese patient who had a clinical diagnosis of Marfan syndrome together with a complex chromosomal rearrangement involving a breakpoint at 3p24.1, disrupting TGFβR2. They also found three missense mutations in four unrelated FBN1-negative Marfan syndrome probands and a splice-site mutation in the original French family, which had mapped to 3p24.2–25.2.
This report led us to reconsider a large family known to us with an autosomal dominant predisposition to aortic dissection, in which no FBN1 mutation was detected in affected members and linkage studies excluded the FBN1 locus.
In 2005, Loeys et al5 reported Loeys–Dietz syndrome, OMIM 609192, consisting of hypertelorism, bifid uvula, cleft palate, arterial tortuosity and ascending aortic aneurysm and dissection due to mutations in TGFβR1 and TGFβR2. They found five missense mutations and a splice-site mutation in TGFβR2 in members from six families, and four missense mutations in TGFβR1 in members from four families with a clinical phenotype indistinguishable from that seen in those with TGFβR2 mutations.
Mutations at the R460 position in TGFβR2 have been reported in four unrelated families with TAAD2,6 and in one family with Marfan syndrome;7 three of the families had the R460H mutation present in our family, and Pannu et al6 proposed that this may be a mutation hotspot.
Recently, there has been a report of a Korean with a de novo missense mutation in TGFβR2, with features consistent with Loeys–Dietz syndrome (fig 1).8
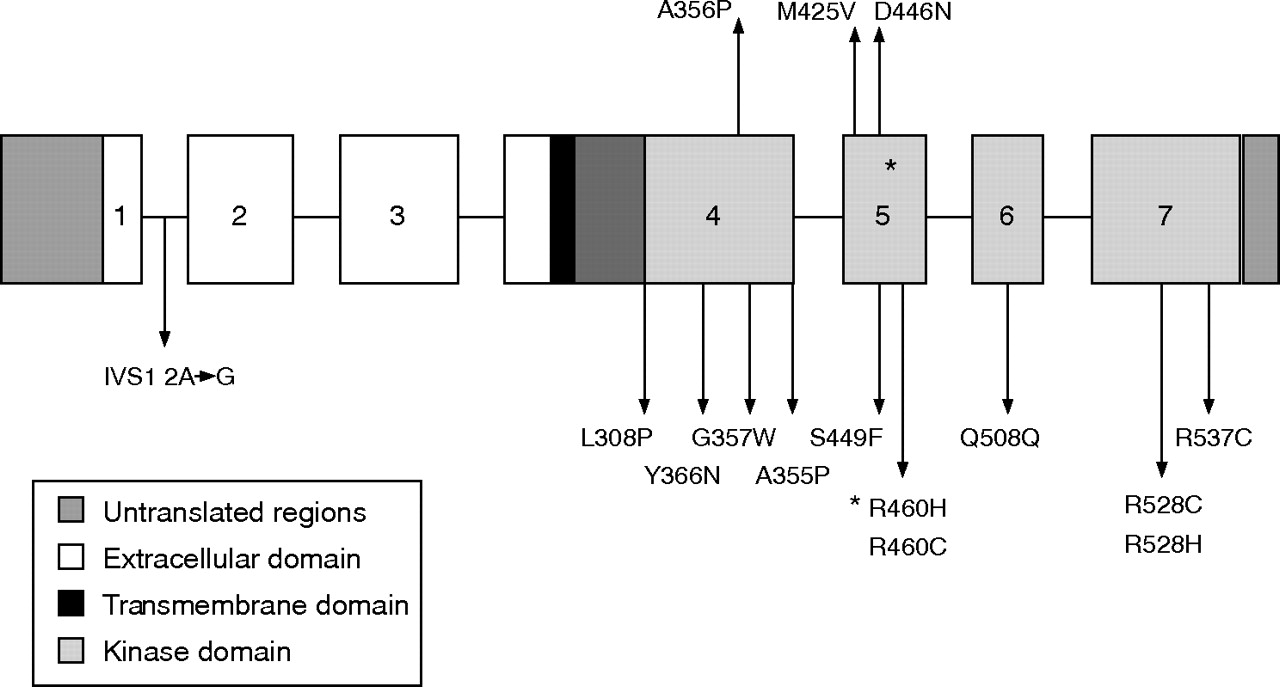
A sketch of transforming growth factor β2 gene with mutations.
TGFBR2, OMIM 190182, codes for a serine threonine kinase transmembrane receptor,9 which, in association with transforming growth factor β receptor 1 and the ligand, transforming growth factor β (TGFβ) is involved in the regulation of cellular processes and the formation of extracellular matrix.10 This pathway may be affected by fibrillin 1, which controls the activation of TGFβ.11 Dysregulation of TGFβ activation has been implicated in the pathogenesis of Marfan syndrome.12–14
METHOD
Patients
Figure 2 shows the family pedigree. There were 107 living members, and there had been eight sudden deaths. Most family members were having periodic echocardigrams. TGFβR2 was sequenced using DNA from an affected member. This showed a missense mutation in exon 5 caused by a G→A substitution, changing amino acid 460 from arginine to histidine. We then offered testing to those individuals at 50% risk of carrying the mutation. At that time, there were nine individuals who were affected on the basis of aortic dilatation or dissection and two obligate carrier mothers, one of whom had a normal cardiac echo and the other aortic and mitral valve regurgitation. These 11 individuals were found to carry the R460H mutation, indicating that the mutation segregated with the disease. The status of the remaining 17 at 50% risk, who elected to be tested, was uncertain; three of these carried the mutation. To date, we have tested 28 individuals and identified 14 with the R460H mutation and 14 without. There were 10 individuals at 50% risk and 9 at 25% risk who have declined testing.

Family pedigree. Written consent was obtained for publication of this image. Circles, females; squares, males; dot inside circle or square, obligate carrier.
All individuals with the R460H mutation, except IV1, were interviewed and examined by CL, and hospital records and investigations such as x rays and ultrasound scans were reviewed.
RESULTS
Table 1 presents the features in each individual.
Features present in individuals
Cardiovascular findings
There were eight sudden deaths between the ages of 27 and 60 years, average age at death 44 years (fig 3). Six individuals had a postmortem examination. The causes of death in these patients were as follows: rupture of dissecting aortic aneurysm at age 55 years; ruptured aortic aneurysm at age 37 years when 39 weeks pregnant; type B aortic dissection (distal to the origin of the left subclavian artery) at age 60 years; ruptured aorta at age 46 years; type A aortic dissection (in the ascending aorta close to the aortic valve) at age 44 years; and type A aortic dissection at age 27 years, (previous subarachnoid haemorrhage at age 26 years). The other sudden deaths, at ages 44 and 41 years, are likely to have been due to aortic dissection based on the accounts of relatives.

Affected family members. Written consent was obtained for publication of this image. d, died; SAH, subarachnoid haemorrhage. Circles, females; squares, males; dot inside circle or square, obligate carrier; numbers, age of individual.
Of the 14 mutation-positive individuals, 8 had a dilated aortic root. The youngest was an 8-year-old boy and the oldest a 67-year-old woman, in whom aortic dilatation had not progressed over the past 10 years; she had also had a subarachnoid haemorrhage at the age of 59 years. Of the eight individuals with aortic dilatation, four had mitral valve regurgitation and of these, two also had mitral valve prolapse. One male family member had an aortic root replacement at age 49 years, and another had an aortic root replacement and mitral valve replacement for significant mitral incompetence at the age of 17 years.
Three female members, aged 34, 33 and 17 years, had a normal aorta on echocardiography, with aortic and mitral valve regurgitation. Two female members, aged 44 and 25 years, had entirely normal echocardiograms. One female member had an apparently normal echocardiogram at the age of 20 years, but 7 months later experienced a type B aortic dissection. This was treated with an endovascular stent. Four months later, she had a further dissection distal to the stent and underwent surgery to replace her descending and abdominal aorta.
Two individuals had a subarachnoid haemorrhage at the ages of 59 and 26 years. Cerebral angiography showed a ruptured berry aneurysm with increased tortuosity of the cerebral vessels in one individual, a subsequent ultrasound scan of the neck showed tortuous carotid arteries. Cerebral angiography, performed some weeks after the acute bleed, in the other individual showed widespread arterial ectasia and unfolding. Three other family members either requested imaging of their cerebral vessels or had this done as part of the investigation for recurrent headaches. All were found to have arterial tortuosity and one had a small berry aneurysm. One individual also had tortuous carotid and vertebral arteries (fig 4).

Tortuous carotid and cerebral arteries and vertebral arteries. Written consent was obtained for publication of this image.
One individual had an aneurysmal brachiocephalic artery, one had an aneurysm of the subclavian artery, and another had a tortuous abdominal aorta.
Of the 22 affected individuals in this family (8 obligate carriers and 14 confirmed mutation carriers), 8 died, 2 required surgery and 1 survived an acute dissection.
Of the 14 mutation carriers, 6 had varicose veins, as did 2 of 3 deceased individuals where records exist. Varicose veins became apparent in the teens in male and female members and recurred after surgery. Some family members developed thread veins in childhood (fig 5).
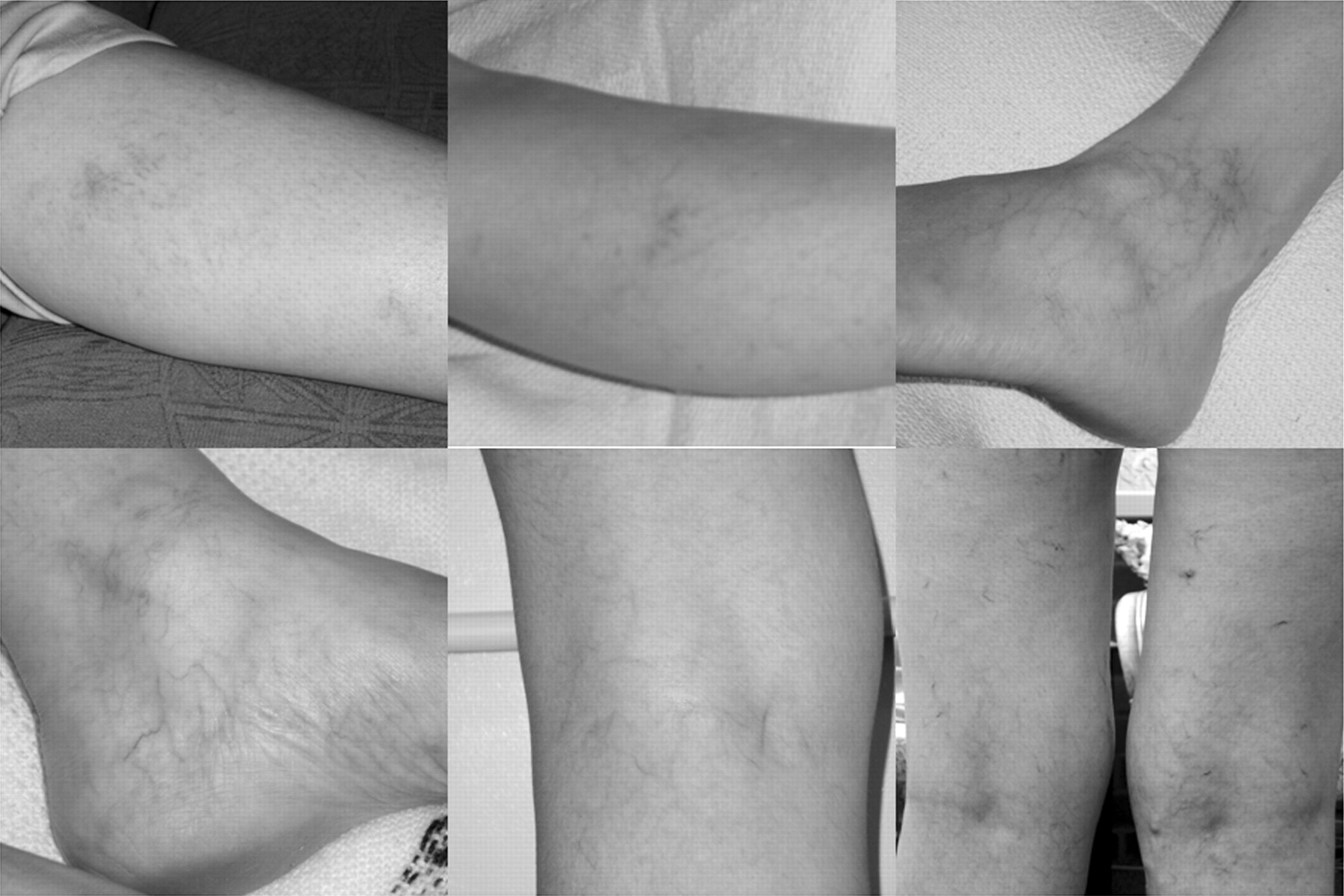
Fine skin with varicose veins and thread veins. Written consent was obtained for publication of this image.
Cutaneous findings
All 14 individuals with the R460H mutation had fine, translucent skin, and 13 of 14 reported easy bruising. Ten of the 14 individuals had striae. These occurred in most of the female members but a young male family member was also affected. They were present on the back and inner thigh in those who had not been pregnant. After pregnancy, individuals were left with florid abdominal striae, which did not diminish with time (fig 6).

Skin striae. Written consent was obtained for publication of this image.
Skeletal and connective tissue findings
The 14 mutation carriers did not have a Marfanoid habitus; 11 had heights between the 9th and 98th centiles; one individual had height at the >99.6th centile and two had heights between the 98th and 99.6th centiles; 13 had a normal armspan:height ratios and no one had arachnodactly, defined as a middle finger length exceeding palm length measured as described by Hall et al15; 7 mutation carriers and 1 of the 3 deceased individuals, for whom a full postmortem report was available had a pectus deformity. Two had a pectus carinatum, four had mild pectus excavatum, and two had asymmetry of the costosternal junction, which was more prominent on one side. Clinically, 3 of 14 carriers and 1 of 3 deceased individuals had a mild scoliosis, with confirmation by x ray in two of the carriers and the deceased individual.
Of the 14, 9 had joint hypermobility defined as a Beighton score16 ⩾5. Individuals tended to become less mobile with age and had pain in multiple joints; five individuals had consulted a rheumatologist. The small joints of the hand were most often involved, with 9 of 14 carriers and 2 of 4 deceased individuals having pain. Six of the 14 carriers and 3 of 4 deceased individuals had back pain or neck pain. Two individuals had undergone a laminectomy, one at the age of 31 years. Other joints involved to a lesser extent were wrists, ankles, knees, hips and elbows. Of those who had been investigated for joint pain, x ray findings were as follows: a female member with degenerative changes in both hip joints, with complete loss of joint space on one side at the age of 62 years, and bilateral degenerative changes at radiocarpal, scaphotrapeziotrapezoid, first carpometocarpal and interphalangeal joints of all fingers at the age of 63 years; a man with retrolisthesis between L5 and the sacrum at the age of 48 years and early degenerative changes in the hip joints at age 52 years; a female with loss of normal cervical lordosis with degenerative changes and loss of disc height between C5 and C6 at the age of 41 years, a female member with degenerative changes in the lower dorsal and upper lumbar spine with osteophytic lipping and marginal sclerosis at the age of 35 years; a female member with bilateral cervical osteoarthritis at the age of 27 years.
Herniae have occurred in 7 of 14 confirmed mutation carriers and in 3 of 4 deceased individuals. The most common type of hernia was inguinal, occurring in six individuals, followed by umbilical in five, and femoral and incisional in one each. Three of ten individuals affected by herniae had herniae at more than one location and three of ten had recurrent inguinal herniae.
Craniofacial findings
Figure 7 illustrates the facial features of the affected individuals.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Facial features of affected family members. Written consent was obtained for publication of this image.
We have collected photographs of all the affected family members except one. Those affected tended to have a long face, a mild facial asymmetry was observed in some family members and palpebral fissures slanting down. Only 1 of the 13 mutation carriers in whom measurement was possible had true hypertelorism, defined as an interpupillary distance >97th centile, measured as described by Hall et al.15 Of the remaining individuals, one had an interpupillary distance <97th centile, one 75–97th centile, six 50–75th centile, two 25th centile and one <25th centile. No one had a cleft palate, but one individual had a bifid uvula. Two had a narrow palate and required dental extractions for dental crowding. The appearance of the uvula and palate in the others were unremarkable.
In profile, some mutation carriers had a prominent nasal root and a convex nose, with a beak-like nasal tip that was more obvious in adulthood.
Ophthalmic findings
Of the 14 mutation carriers, 4 reported having treatment for strabismus in childhood. None of the 14 had lens dislocation. Of the five mutation carriers who had a detailed eye examination by an ophthalmologist, none had an abnormally flat cornea, hypoplastic iris or ciliary muscles causing miosis. One, aged 27 years, had myopia with a visual acuity of 6/9 in the right eye and 6/6 in the left. This individual had a right posterior vitreous detachment, with no evidence of lattice degeneration. The second, aged 26 years, had a left esotropia, hypermetropia and amblyopia; the third, aged 52 years, had esotropia and mild amblyopia; the fourth, aged 8 years, had mild hypermetropia; and the fifth, aged 44 years, had minor esotropia.
OTHER FEATURES
Migrainous headache
Of the 14 mutation carriers, 11 had recurrent migrainous headaches, which often started in childhood.
Excess fatigue
Of the 14 mutation carriers, 11 complained of what they perceived to be increased fatigue; they could not keep up with their peers and had daytime somnolence.
DISCUSSION
To date, clinical information has been published about members from 18 families with a pathogenic mutation in TGFβR2,4–8 all of which have been missense or splice-site mutations. Of these families, five6,7 had a change of amino acid at R460, and, of these, three6,7 had the same R460H mutation as in the family we describe.
TGFβR2 mutations have been found in association with Loeys–Dietz syndrome,5,8 Marfan syndrome,4,7 a Marfan-like disorder4,7 and TAAD2.6
The family we describe had a striking cardiovascular phenotype, the cardinal feature being a high risk of aortic dissection, both type A and type B, which can occur at aortic root diameters less than those considered to require surgery in Marfan syndrome.17 Individuals also had a generalised vasculopathy, giving rise to arterial tortuosity and arterial aneurysms and in particular intracranial aneurysms leading to subarachnoid haemorrhage. These findings are similar to those described by others.5,6,8
No family member had ocular lens dislocation, and no ocular features of Marfan syndrome were found in those examined by an ophthalmologist. Lens dislocation has been described in one individual, MS1-IV-83,4 with a TGFβR2 mutation, but there is little clinical information about the nature of the lens dislocation in this individual and lens dislocation was not present in any other affected member of the family. Lens dislocation has not been described in any other individual with a TGFβR2 or TGFβR1 mutation.1,3–8 Most individuals examined were not tall, and did not have dolichostenomelia or arachnodactyly. All patients had translucent, fine skin and most reported easy bruising. This combination of features is not characteristic of Marfan syndrome. However, some individuals did have features such as scoliosis and pectus but to a milder degree than usually seen in Marfan syndrome; joint hypermobility, herniae and striae were common.
The Ghent criteria18 were applied to the 13 individuals who were interviewed and examined. Five had a slit-lamp examination by an ophthalmologist, and it was not possible to investigate for lumbosacral dural ectasia. In five, the skin was involved, due to the presence of striae. Finding lumbosacral dural ectasia in these individuals would not have allowed them to fulfil the Ghent diagnostic criteria for Marfan syndrome. Two met one major criterion, aortic root dilatation, but they did not have involvement of another system, so the presence of lumbosacral dural ectasia would not have been enough to make a diagnosis of Marfan syndrome. Six individuals fulfilled a major criterion; in all cases, this was because of aortic root dilatation. In five of these, the skin was involved due to the presence of herniae and or striae, and one had skeletal involvement, with a positive wrist and thumb sign, joint hypermobility and a high-arched palate with dental crowding. These would have met the Ghent criteria if lumbosacral dural ectasia was present. However, the phenotype in these individuals is sufficiently distinct from Marfan syndrome to distinguish their condition as a different and separate entity. To date, one patient with a TGFβR2 mutation has been reported with lumbosacral dural ectasia5; it was not present when sought in other cases,3–5,7,8 suggesting that this may not be a common finding.
Members of this family did have many of the clinical features of Loeys–Dietz syndrome,5 in particular, aortic root dilatation, aortic dissection, mitral valve prolapse, arterial tortuosity and arterial aneurysms, scoliosis, pectus, joint hypermobility and fine skin. However, not all features described in Loeys–Dietz syndrome were present in our patients. No one had a bicuspid aortic valve, patent ductus arteriosis or bicuspid pulmonary valve. Hypertelorism and bifid uvula were not as prevalent, and none of the members in this family had any learning difficulties or developmental delay. Two of the members reported by Loeys had craniosynostosis,5 but this was not present in five others with Loeys–Dietz syndrome due to a TGFβR2 mutation5,8 and was not a feature in this family. It is interesting that craniosynostosis was not present in the individual with an R460H mutation described by Disabella et al,7 nor in those with TAAD2.6 It is known that tgfbr2 in the mouse is involved in craniofacial development,19–21 and it is possible that the facial asymmetry present in some members of this family could represent a milder result of interference with the same developmental process, which results in craniosynostosis in its most extreme form. Alternatively, it is possible that the R460H mutation in this family does not affect craniofacial development to the same extent as the G357W and G95-2 A→G mutations,5 or that other modifying factors in this family ameliorated the affects of the R469H mutation on craniofacial development.
Striae and herniae are not described in Loeys–Dietz syndrome, but were common in members of this family; indeed, striae4,7 and herniae3,4 have been reported in other individuals with TGFβR2 mutations. Varicose veins, migrainous headaches and excessive fatigue have not been described before in others with a TGFβR2 mutation. The presence of these in members of this family suggests that they may be features of a TGFβR2 mutation.
The cardiovascular findings in family members are remarkably similar to those described in the four TAAD2 families with TGFβR2 R460 mutations.6 In these families, it was suggested that the penetrance of cardiac complications may be reduced in female members. Our family does not support this, but there is an over-representation of affected female members, 17 compared with 5 male family members. The clinical information pertaining to these individuals6 concentrates on the cardiovascular findings. There is no suggestion that they had involvement of other systems. This contrasts with the clinical findings in this family and the other reported case with an R460H mutation,7 which would suggest that the R460H mutation causes a more extensive connective tissue disorder, manifested by striae, herniae and joint hypermobility, of variable expression and penetrance. However, many of the features in our family, such as fine, translucent skin, striae, varicose veins, hernias and joint hypermobility, could be overlooked unless specifically sought. It was only by examining the 13 family members that a clearer picture of the phenotype emerged. It would be interesting to know, in the light of the findings in this family, if other individuals with the same R460H mutation have a similar phenotype.
It is noteworthy that, with the exception of one individual, no one with a TGFβR2 mutation has been reported to have ocular lens dislocation. Studies in mice22 have shown that the protein TGFβ2, but not TGFβ1 or TGFβ3, is expressed in the developing lens and that Tgfbr2 is expressed in the periocular mesenchyme, lens, retina and primary vitreous. Mice with a homozygotic deletion of exon 4 of Tgfbr2 showed microphthalmia and anterior chamber anomalies similar to those seen in Axenfeld–Rieger anomaly, together with defects of the posterior segment.22 Mice heterozygotic for the deletion or with wild-type Tgfbr2 showed normal eye development.22 It would seem that in humans with a TGFβR2 mutation, there is sufficient TGFβ2 signalling to allow normal eye development. Although dysregulation of TGFβ is clearly involved in the pathogenesis of Marfan syndrome,12–14 there must be factors and mechanisms other than the TGFβ pathway to account for the different eye findings in those with FBN1 mutations and TGFβR2 mutations. A study of the interactions of the fibrillin and TGFβ pathways in the eye may help to determine if Marfan syndrome and Loeys–Dietz syndrome arise from events occurring at different places in a single developmental pathway or due are to events in entirely different developmental processes.
We believe that the R460H mutation in TGFβR2 in this family causes a condition that is clinically distinct from Marfan syndrome. The risk of aortic dissection seemed greater than in Marfan syndrome and occurred at smaller aortic root diameters. Arterial tortuosity and aneurysms were common and present in all those investigated. There was no ocular involvement characteristic of Marfan syndrome, and individuals had fewer skeletal features than seen in Marfan syndrome. Affected family members also had fine, translucent skin that was easily bruised, which is not a feature of Marfan syndrome.
Although the cardiovascular findings in the family were consistent with TAAD2, the family also had a systemic connective tissue disorder, which distinguishes their condition from TAAD2. The clinical findings in this family lead us to believe that mutations in TGFβR2 cause a different disorder from that caused by mutations in FBN1. The other cases reported with mutations in TGFβR2 do have common features. The differences could conceivably have resulted from clinicians focusing on different aspects of a single entity with a variable and subtle phenotype and variable penetrance.
We would advocate TGFβR2 and TGFβR1 mutation testing in any individual with clinical features similar to those described, particularly if there is a family history of aortic dissection, although this is not mandatory, as de novo mutations are described. individuals who are thought to have Marfan syndrome, in whom no FBN1 mutation has been found, with no personal or family history of lens dislocation should also be considered for testing.
Mutation testing in this family has allowed us to identify those at risk and has enabled us to reassure 68 individuals and cease their annual cardiac screening.
The findings in the 14 confirmed mutation carriers have implications for the cardiac screening of individuals with a TGFβR2 mutation. We would suggest that cardiac screening should start in childhood. The youngest child with a dilated aorta in this family was an 8-year-old boy. Screening should be regular, six monthly seems appropriate, and lifelong for mutation carriers and those at 50% risk. However, a normal echocardiogram cannot be totally reassuring and other screening modalities such as magnetic resonance imaging may be more appropriate. The entire aorta is at risk of dissection and consequently requires imaging, and individuals continue to be at risk of dissection elsewhere in the aorta after replacement surgery. Prophylactic surgery should be considered at diameters less than those seen in Marfan syndrome. Pregnancy is a time of increased risk for women with a TGFβR2 mutation, as seen in individual II1 and therefore we would advise extra vigilance at this time.
Arterial aneurysms, in particular intracranial aneurysm leading to subarachnoid haemorrhage, seem to be a consistent finding in individuals with a TGFβR2 mutation,5,6 although the absolute risk of subarachnoid haemorrhage seems to be lower than that of aortic dissection. It is not yet clear whether screening for intracranial arterial tortuosity and aneurysms is beneficial to individuals, but this may become clearer as more families are reported and the natural history is better defined.
Acknowledgments
We thank all family members for their determination to solve the mystery of their family history and their constant cooperation. We also thank Dr Eli Hatchwell for his contribution and Dr Nick Dennis for his comments.
REFERENCES
Footnotes
-
Published Online First 2 August 2006
-
Competing interests: None.
-
Written consent to publish clinical information and photographs was obtained from all living family members or from parents of children. Verbal consent was obtained from next of kin in the case of deceased individuals.