Article Text

Abstract
Schizophrenia is a severe mental disorder affecting approximately 1% of the world’s population. Although the aetiology of schizophrenia is complex and multifactorial, with estimated heritabilities as high as 80%, genetic factors are the most compelling. Childhood-onset schizophrenia (COS), defined as onset of schizophrenia before the age of 13 years, is a rare and malignant form of the illness that may have more salient genetic influence. The first known case of paternal segmental uniparental isodisomy (iUPD) on 5q32-qter in a patient with COS is described, which adds to the previously known high rates of chromosomal abnormalities reported in this sample. iUPD is a rare genetic condition in which the offspring receives two chromosomal homologues from one parent. Segmental UPD is defined as UPD on a portion of a chromosome with biparental inheritance seen in the rest of the homologous pair. Complications owing to this abnormality may arise from malfunctioning imprinted genes or homozygosity of recessive disease-causing mutations. This aberration became apparent during whole-genomic screening of a COS cohort and is of particular interest because 5q has been implicated in schizophrenia by several genomewide linkage studies and positive gene associations. This report, therefore, presents more evidence that schizophrenia susceptibility gene, or genes, may be found on distal 5q.
- COS, childhood-onset schizophrenia
- DSM, diagnostic manual for mental disorders
- GABA, γ-aminobutyric acid
- iUPD, uniparental isodisomy
- MRI, magnetic resonance imaging
- PCR, polymerase chain reaction
- SNP, single-nucleotide polymorphism
- UPD, uniparental disomy
Statistics from Altmetric.com
- COS, childhood-onset schizophrenia
- DSM, diagnostic manual for mental disorders
- GABA, γ-aminobutyric acid
- iUPD, uniparental isodisomy
- MRI, magnetic resonance imaging
- PCR, polymerase chain reaction
- SNP, single-nucleotide polymorphism
- UPD, uniparental disomy
Schizophrenia is a severe mental disorder affecting approximately 1% of the population worldwide. Many factors are believed to contribute to the aetiology of the illness, but given the high heritability rates (>80%),1 genetic vulnerability is currently the most compelling factor being examined. Childhood-onset schizophrenia (COS), defined as onset of schizophrenia before the age of 13 years, is a rare and malignant form of the illness that may have more salient genetic influence.2,3
Uniparental disomy (UPD) is a genetic condition in which the offspring receives two chromosomal homologues from a single parent. The most commonly reported occurrence of UPD occurs on chromosome 15 and causes Prader–Willi or Angelman’s syndrome. If the UPD is present on a portion of the homologues, the condition is referred to as a segmental UPD, and if the homologues are identical (copies of a single homologue from one of the parents), the condition is known as isodisomy. Segmental isodisomies with a normal karyotype are rarely reported and occur by complex mechanisms. One suggested mechanism begins with recombination in a tetrad that is followed by non-disjunction in meiosis I. This leads to a germ cell with a diploid chromosome that can be fertilised to create a trisomic zygote. This is followed by mitotic crossing-over between a chromosome from each of the parents and an event termed trisomy rescue, in which one of the three homologues is lost to create a seemingly normal, diploid cell.4 UPD is a rare condition, and to date there has been only one report of a whole-chromosome UPD on chromosome 5 in a child with spinal muscular atrophy.5,6
Key points
-
This is the first report of paternal segmental uniparental isodisomy on 5q32-qter in a patient with childhood-onset schizophrenia (COS).
-
COS has a high rate of chromosomal abnormalities.
-
The availability of high-density microarray technologies provides opportunity to discover rare genetic events such as uniparental disomies
The increasing availability of whole-genome scans has provided a more effective means to support and discover rare genetic events such as UPD. We hypothesised that given the extreme severity of the illness in the COS cohort, they would be an ideal population to scan for rare genomic aetiologies using multiple complementary methods, including high-density comparative genomic hybridisation, single-nucleotide polymorphisms (SNPs) and expression microarrays. Here, we describe the first reported case of a paternal segmental uniparental isodisomy on 5q32-qter.
METHODS
Patient recruitment and clinical assessment
Patients meeting the Diagnostic and statistical manual of mental disorders—third edition revised/Diagnostic and statistical manual of mental disorders—fourth edition (DSM-III-R/DSM-IV)7 criteria for schizophrenia or psychosis not otherwise specified were recruited nationwide through an extensive screening process, including a review of more than 1400 charts and in-person screening of >230 subjects. The National Institute of Mental Health Institutional Review Board approved the project and written consent was obtained from parents and assent from minor subjects. The patients who participated were required to have a premorbid full-scale IQ of ⩾70 and an onset of psychotic symptoms before the age of 13 years. The diagnosis of COS was confirmed by two psychiatrists who achieved good reliability as measured by κ8 = 0.8 for the first 19 cases diagnosed9 through an extensive evaluation that included clinical and structured interviews of the children and parents using portions of the Schedule for Affective Disorders and Schizophrenia for School-Age Children10,11 and in-hospital observation during a 1 to 3-week drug-free period. Information on these subjects included cognitive and behavioural ratings of early development, history of drug response, neuropsychological test performance, smooth pursuit eye movements and magnetic resonance images (MRIs).
Proband
The female patient is the first-born of a physically healthy and unrelated couple. She has a healthy brother who is 2 years younger. The patient was breech, but birth was by a planned caesarean section; pregnancy and delivery were otherwise unremarkable. The baby was healthy, and the child reached developmental milestones within normal ranges. No unusual behaviour or problems were noted until the child began school and teachers remarked that she was inattentive and internally preoccupied. At the age of 9 years, her parents noticed avoidant and isolative behaviour, as well as deterioration in hygiene. She was hospitalised during an episode in which she refused to eat and was diagnosed with atypical psychosis and general anxiety disorder. By the age of 11 years she was frankly psychotic, experiencing auditory hallucinations, delusions and an increasing lack of drive and affective flattening.
The patient was screened at the National Institute of Mental Health at 12 years of age, and during the interview she was agitated, actively avoided eye contact with the interviewer, was neologistic and seemed to be laughing and conversing with herself. She was experiencing psychotic symptoms including delusions of reference, and concern with punishment from god and was hearing voices. The interview disclosed anhedonia, as well as impairment in school, relationships and self-care. She had no unusual physical or neurological features and was fully oriented. Neuropsychometric assessment was difficult owing to her mental state, which was disclosed by her scores on the Wechsler Intelligence Scale for Children—Revised of 45 and 46 for verbal and performance IQ, respectively. This was in contrast with a report from her first hospitalisation, which stated “given her average test scores on three of the subtests, it is hypothesised by this examiner that the patient’s abilities are at least in the average range of intelligence”. Unfortunately, actual scores were not reported in her medical records, only a summary statement from the psychologist who administered the test. Also, she was successful in attending regular public school until the time of her first hospitalisation. Standard laboratory tests were carried out, including electroencephalography, MRI and a cytogenetic karyotype on peripheral blood, all of which were normal. She met DSM-III criteria for axis 1 schizophrenia and major depression, and criteria for an axis II learning disability.
The patient was treated, as an outpatient, with olanzapine and experienced adverse effects caused by the drug. After a failure to respond to multiple other antipsychotics, the patient was treated with clozapine, which produced a moderate, sustained clinical and functional improvement, noted at follow-up when she was 14 years old.
Proband family history
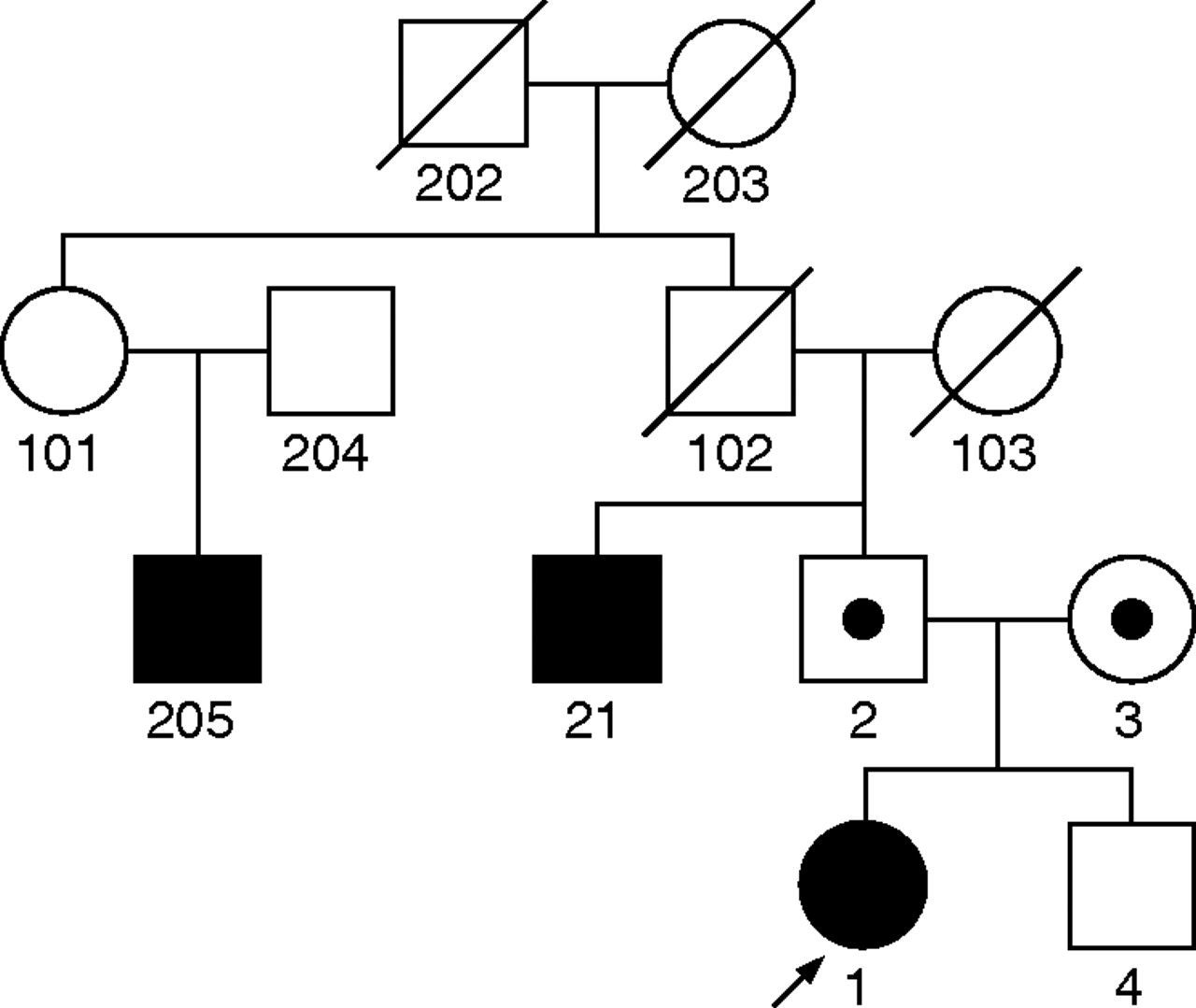
The patient’s family exhibited several disorders. Her mother met DSM-III criteria for axis II histrionic and schizoid personality disorders. The father was diagnosed with axis I major depression and social phobia, as well as axis II schizoid, schizotypal and avoidant personality disorders. The brother, evaluated at the age of 10 years, was found to be mentally healthy and was experiencing no known problems at home or school. His healthy status was confirmed again during a follow-up interview 3 years later. Further, on the basis of proxy information from the parents, the patient had a paternal uncle and paternal second cousin diagnosed with schizophrenia. The patient also has a paternal second cousin diagnosed with bipolar disorder (fig 1, created with Pelican software12).

Family affected by schizophrenia and schizophrenia spectrum disorders, including proband with uniparental disomy.
DNA preparation and genotyping
Genomic DNA was extracted from immortalised lymphoblastoid cells, using the QIAamp DNA Extraction Kit (Qiagen, Valencia, California, USA). Several complementary genomic scans were completed. Firstly, a genomewide microsatellite markers scan with semiautomated fluorescent genotyping was undertaken using the ABI Genescan/Genotyper system (Applied Biosystems, Foster City, California, USA) by comparison of the fragment sizes with an internal standard (LIZ). A total of 21 microsatellite markers on chromosome 5 with an average distance between adjacent markers of 9.6 cM (2–28 cm) and an average heterozygosity of 0.80 (0.73–0.89) were selected from the ABI prism Linkage Mapping Set, V.2.5. Multiplex polymerase chain reaction (PCR) tests were carried out with 10 ng of genomic DNA and 9 μl of True Allele PCR Premix (Applied Biosystems). The PCR amplifications were set up on a Biomek FX robotic workstation (Beckman Coulter, Fullerton, California, USA) and run on MSB 0.25 Thermo Hybaid PCR machines (Franklin, Massachusetts, USA). Cycling conditions were as follows: initial denaturation at 95°C for 12 min, followed by 30 cycles of denaturation at 94°C for 15 s, annealing at 55°C for 15 s and extension at 72°C for 30 s. The PCR products were then pooled and the internal size standard (LIZ) was added. The alleles were detected by electrophoresis on the ABI 3700 DNA Analyzer. The software Genescan and Genotyper (Applied Biosystems) were used to size and call the alleles. All genotypes were scored blind as to phenotype.
Secondly, samples were processed according to the Affymetrix GeneChip Mapping 500K Assay Manual using the Mendel_NSP1_EA arrays13 as part of the early access programme through the UCLA site of the National Institutes of Health Neuroscience Microarray Consortium. Genotypes were called in GTYPE software (Affymetrix, Santa Clara, California, USA). Intensity data were analysed in custom scripts in Mathematica (B Merriman).
Microarray gene expression sample preparation
Total RNA was extracted from lymphoblastoid cell lines using a Trizol protocol available from the manufacturer (Invitrogen, Carlsbad, California, USA). RNA (500 ng) was used with the Illumina Total Prep RNA Amplication Kit (Ambion, Austin, Texas, USA) to prepare RNA with incorporated Biotin-16-UTP (Perkin Elmer, Wellesley, California, USA). cRNA yields were quantified using Invitrogen’s RiboGreen, and 1.5 μg of the biotinylated cRNA was hybridised to a Sentrix Human-6 Expression Beadchip (Illumina, San Diego, California, USA), which contained approximately 48 000 known and predicted genes. Washing and scanning were carried out according to the manufacturer’s protocol, and a detailed description of the BeadChip system has been provided elsewhere.14 Samples were coded and run in duplicates, and the results were analysed using the rank invariant normalisation method in the BeadStudio analysis software (http://www.illumina.com).
Candidate gene SNP genotyping and transmission disequilibrium test
To follow-up on potential candidate genes in the affected region on 5q, we selected SNPs from previous publications of positive associations with schizophrenia to test for association in the entire COS cohort (n = 88) and their parents (n = 150). Primers and probes were ordered through Assays by Design (Applied Biosystems) for the TaqMan Assay, and we used standard PCR protocols and read plates on the Prism 7900HT. TDTPhase was used to compute phase-known transmission disequilibrium test associations.15
RESULTS
A comparison of the proband’s cognitive abilities as well as volumetric MRIs of the brain indicated that she was among the most extremely affected in our already severely affected cohort of patients with COS. Specifically, her IQ score on admission to the National Institutes of Health was 40, which was the lowest of any of our patients, with only a 7-point improvement at the 3-year follow-up. Further, her overall grey matter was among the smallest in comparison with the other female patients with COS, ranking 13–15 of 15 in every lobe of the brain (details available on request).
The Affymetrix 250K Mendel_Nsp_EA SNP array showed a 35-Mb stretch of homozygosity from 5q32-qter, which encompassed 3400 SNPs. Intensity data indicated that the large interval was diploid as comparable intensity signals were seen across a series of normal diploid controls. Thus, the aberrantly long stretch of homozygosity was not due to chromosomal loss. This was consistent with the normal karyotype results. Therefore, the loss of heterozygosity was most probably attributable to an iUPD.
Multiallelic microsatellite markers, over the region-of-interest defined by the SNP arrays, showed misinheritance at 3 of 4 markers on 5q (fourth marker was uninformative). All questionable genotypes were repeated to rule out genotyping errors. In all cases, the proband apparently inherited an allele from the father, but none from the mother. Genotypes of the healthy sibling were fine (fig 2). Thus, the isodisomic region was paternally inherited. This region is of particular interest because this region of chromosome 5 overlaps with a region implicated in schizophrenia by genome-scan linkage meta-analysis studies.16,17 Further, positive associations of SNP alleles in several γ-aminobutyric acid (GABA) receptor genes in the region have been reported.18 Therefore, we attempted to replicate the reported positive SNP associations by using transmission/disequilibrium test in the entire COS cohort, but none of the SNPs tested showed any association with COS (table 1).
Transmission disequilibrium test results of single-nulceotide polymorphisms in candidate genes in the 5q region

{kind=link}
{kind=link}
Inheritance errors in affected family using microsatellite markers.
The Sentrix Human-6 Expression Beadchip contains approximately 300 probes corresponding to 253 known and predicted genes in the 35-Mb-affected region on 5q. A direct comparison of expression levels of these genes between the proband and her unaffected brother showed 8 genes that were up regulated and 21 genes that were down regulated, with fold changes ranging from 1.3 to 17 (table 2). Of the genes identified, none were obvious candidates. Unfortunately, we were not able to detect any of the GABA receptor genes in this region at reliable levels, which reflects one of the inherent limitations of using mRNA from lymphocytes rather than brain tissue.
Significantly differentially expressed genes on 5q in lymphoblasts between proband and healthy sibling
DISCUSSION
We used high-density SNP arrays to reliably search through the genome of people with COS and discovered a paternal segmental iUPD on 5q32-qter in a single patient. To date, only one other case of UPD on chromosome 5 has been reported.5,6 This report, therefore, supports a location of some schizophrenia susceptibility gene, or genes, in 5q, as well as provides what may be a novel region of known imprinting. Further, it adds to the body of evidence indicating that the extreme cohort of patients with very early onset of schizophrenia harbour a large number of rare chromosomal abnormalities that are probably associated with the aetiology.
In a 2003 weighted meta-analysis, 5q23.2−q34 was ranked as the second most compelling linkage region for schizophrenia.16 Subsequent to that meta-analysis, Sklar et al17 found a region on 5q31–5q35 with a non-parametric Linkage Score of 3.28 in a Portuguese sample and successfully replicated the findings in a bipolar population in an attempt to connect it to a psychotic phenotype.17 Further study of this region showed positive SNP associations with several GABA receptor subunit genes, which are plausible candidates based on prior evidence for GABA system involvement in schizophrenia, in Portuguese and German patients with schizophrenia.18 Pimm et al19 reported an association with the Epsin 4 gene found on 5q33.3, in a large case–control sample from the UK, and this gene was further implicated by a follow-up study on a Chinese sample that showed an association between Epsin 4 haplotypes and schizophrenia.20 This gene is a schizophrenia candidate owing to its involvement with neurotransmitter vesicle transport and stability in the synapses and neurones.19 Taken together, these studies provide strong evidence for schizophrenia susceptibility in the region in which we have confirmed an iUPD, a chromosomal abnormality that can be detrimental owing to the homozygosity of autosomal recessively inherited disorders and imprinting.
Imprinting occurs when expression levels are altered due to gene modifications determined by the parent of origin.21 We determined that the isodisomy in the proband is paternal, which suggests that paternally imprinted genes in this region will lack expression. Imprinting maps are currently being developed for the human genome, and although there is no conclusive evidence for imprinting in this region, recent projects suggest it is a strong possibility. An expression study using FANTOM2 mouse complementary DNA clones suggests that up to 2101 of 27 663 (7.6%) investigated transcripts are imprinted. The transcripts were mapped to the human genome, and several of the suggested imprinted genes are found in the region of the isodisomy concerned.22 Luedi et al21 developed a more conservative model and suggest that 2.5% of murine genes are imprinted, 64% of which exhibit maternal expression. Human-based literature also supports the likelihood of imprinted genes in this region, such as research on the SPINK5 gene found on 5q32, which shows disease association with the maternally derived allele.23 This research is encouraging regardless of the gene involvement with schizophrenia because imprinted genes typically cluster together.22
Also of interest is the use of the proband’s genome to pinpoint candidate genes, which is possible given the specifics of the isodisomy and the patient’s family history. Although both parents of the proband meet criteria for schizophrenia-spectrum disorders, the paternal side of the family presents a history of schizophrenia, which is absent on the maternal side. The isodisomy is paternal, so the father could be a source of a homozygotic recessive mutation responsible for illness in the patient. If the father was heterozygotic for these variants, there is a possible explanation for his meeting criteria for a schizophrenia-spectrum disorder rather than schizophrenia. Full sequencing of the entire region in the proband and her family may be the only way to identify potential disease-causing mutations. Although the possibility of disease contributions lying outside chromosome 5 cannot be ignored, the implications brought about by the family history could implicate a monogenic form of this complex disease.
Although linkage studies have been successful in identifying a few schizophrenia candidate genes, the discovery of a specific chromosomal abnormality in broad linkage regions may be helpful in isolating a specific locus.24 Such abnormalities include the 22q11 deletion and the balanced translocation of chromosome 1, which have provided the strongest genetic predictors of schizophrenia and psychosis.24,25 Breakpoint analysis in a segmental isodisomy may also implicate novel genes in the study of schizophrenia. In a 2001 case report, Kotzot et al26 discussed mechanisms by which a segmental isodisomy is created when a chromosomal breakage occurs in mitosis and is followed by reduplication. In this case, mitotic reduplication occurs to compensate for the loss by making a replacement segment from the intact homologue. The same group has also proposed mechanisms that involve recombination in meiosis and mitosis that may be responsible for a segmental isodisomy.4 These mechanisms provide scenarios in which breakage could have a direct effect on a specific gene. Parental age may also have a role in the occurrence of isodisomies.27 It is noteworthy that the parents of the proband were among the oldest in our cohort at 41 and 37 years of age when she was born.
SUMMARY
Utilisation of high-density SNP arrays has the potential to expose a considerable amount of previously unknown information regarding rates of iUPDs. Prior detection of this genetic condition has relied on time-consuming analysis using microsatellite markers and, as a result, most reports to date have been serendipitous.4 Experiments testing the effectiveness of arrays in identifying uniparental disomies seem promising,28 and along with other chromosomal abnormalities, isodisomies will probably be key in developing imprinting maps and discovering and confirming regions of risk in many disorders. This case report supports this notion as well as the possibility of finding risk genes and imprinting on 5q.
Acknowledgments
We thank all the patients and their families who have participated in our study.
REFERENCES
Footnotes
-
Published Online First 8 June 2006
-
Competing interests: None declared.