Article Text

Abstract
Background: Autism spectrum disorders (ASD) refer to a broader group of neurobiological conditions, pervasive developmental disorders. They are characterised by a symptomatic triad associated with qualitative changes in social interactions, defect in communication abilities, and repetitive and stereotyped interests and activities. ASD is prevalent in 1 to 3 per 1000 people. Despite several arguments for a strong genetic contribution, the molecular basis of a most cases remains unexplained. About 5% of patients with autism have a chromosome abnormality visible with cytogenetic methods. The most frequent are 15q11–q13 duplication, 2q37 and 22q13.3 deletions. Many other chromosomal imbalances have been described. However, most of them remain undetectable using routine karyotype analysis, thus impeding diagnosis and genetic counselling.
Methods and results: 29 patients presenting with syndromic ASD were investigated using a DNA microarray constructed from large insert clones spaced at approximately 1 Mb intervals across the genome. Eight clinically relevant rearrangements were identified in 8 (27.5%) patients: six deletions and two duplications. Altered segments ranged in size from 1.4 to 16 Mb (2–19 clones). No recurrent abnormality was identified.
Conclusion: These results clearly show that array comparative genomic hybridisation should be considered to be an essential aspect of the genetic analysis of patients with syndromic ASD. Moreover, besides their importance for diagnosis and genetic counselling, they may allow the delineation of new contiguous gene syndromes associated with ASD. Finally, the detailed molecular analysis of the rearranged regions may pave the way for the identification of new ASD genes.
- array-CGH, array-based CGH
- ASD, autism spectrum disorders
- CGH, comparative genomic hybridisation
- DSM IV, Diagnostic Statistical Manual for Mental Disorders—fourth edition
- FISH, fluorescence in situ hybridisation
- PDD, pervasive developmental disorder
- PDD—NOS, PDD—not otherwise specified
Statistics from Altmetric.com
- array-CGH, array-based CGH
- ASD, autism spectrum disorders
- CGH, comparative genomic hybridisation
- DSM IV, Diagnostic Statistical Manual for Mental Disorders—fourth edition
- FISH, fluorescence in situ hybridisation
- PDD, pervasive developmental disorder
- PDD—NOS, PDD—not otherwise specified
Autism spectrum disorders (ASD) belong to the group of pervasive developmental disorders (PDD). According to the Diagnostic statistical manual for mental disorders—fourth edition (DSM IV) classification,1 ASD are characterised by impairments in communication, social skills and restricted or stereotyped pattern of behaviours and interests. A diagnosis within the autism spectrum requires one or more symptoms in each of the three areas of impairment. The prevalence of ASD is estimated at about 1/1000 to 3/1000.2,3 ASD are heterogeneous conditions which can be either isolated or syndromic—that is, associated with other clinical features such as facial dysmorphism, limb or visceral malformations, and growth abnormalities.
A total of 10–20% of ASD cases are due to known medical conditions involving chromosomal imbalances, genetic disorders (X fragile syndrome and tuberous sclerosis)4 or environmental factors (valproate5 and rubella). The other cases remain unexplained. Twin and familial studies have documented a higher concordance rate in monozygotic twins (90%) than in dizygotic twins (4.5%),6–8 and a 75-fold greater risk to siblings in idiopathic patients than in the general population.9,10 Collectively, these studies support the involvement of numerous genes in autistic disorders.
About 1.7–4.8% of people with ASD have chromosome abnormalities. Almost all chromosomes have been involved, including unbalanced translocations, inversions, rings, and interstitial or terminal deletions and duplications.11–14 The rare chromosome abnormalities that have been reported on more than one occasion are duplication of 15q,15 deletions of 18q,16,17 Xp,18,19 2q37,20 22q1321,22 and the sex chromosome aneuploidies 47,XYY23,24 and 45,X/46,XY.25,26 This diversity of loci suggests that studying chromosomal aberrations in relationship to autism will require efficient and highly sensitive tools. In addition to the importance for diagnosis, identification of chromosomal imbalances in patients with ASD may also be instrumental for cloning disease-causing genes. Analysis of Xp22.3 deletion has indeed allowed the identification of the NLGN4 gene.27
Recent technological developments, such as array-based comparative genomic hybridisation (array-CGH),28–30 allow the investigation of the human genome at a resolution that is 5–10 times higher than that of routine chromosome analysis by karyotyping.29,31–33 Array-CGH has been used successfully for analysis of tumour samples and cell lines, and for high-resolution analysis of patients with mental retardation and congenital anomalies.34–38
Here, we report the application of genomewide array-CGH, at 1 Mb resolution, to the study of 29 patients with syndromic ASD. In addition to their clinical relevance, our results emphasise the importance of chromosomal imbalance in the aetiology of syndromic ASD and may help the identification of new genes involved in autistic disorders.
METHODS
Participants
We recruited patients born to unrelated parents and presenting with a syndromic ASD. All patients fulfil the DSM IV criteria for autism, PDD—not otherwise specified (PDD—NOS) or Asperger’s syndrome. Twenty eight patients were selected consecutively by clinical geneticists or child psychiatricians of the Necker-Enfants Malades Hospital in Paris, France. One patient was recruited from the PARIS study (Paris Autism Research International Sibpair study).39 All patients were evaluated by a clinical geneticist and most of them had psychometric tests carried out by a psychologist. In addition, the results of high-resolution karyotype (approximately 800 bands), biochemical tests and haematological examinations were normal in all patients.
Blood samples were obtained from the probands and their parents after informed consent, and genomic DNA was isolated from blood leucocytes using the Nucleon kit (Amersham, Buckinghamshire, UK) according to the manufacturer’s instructions.
Array-CGH
The 1-Mb resolution arrays used in this study are as described previously.40 Arrays developed by the Wellcome Trust Sanger Institute (Hinxton, Cambridge, UK) are made of 3523 clones (BAC or PAC, size between 80 and 240 kb). These clones are spread over the entire genome with a mean resolution of 1 clone/Mb. Subtelomeric regions as well as most characterised microdeletion syndromes are included in these clones. All clones are set in duplicate.
Array hybridisation using the 1 Mb array was undertaken as described previously.40 Briefly, Cy3 and Cy5-labelled DNAs were combined, precipitated with 135 μg of human Cot1 DNA (Roche, Mannheim, Germany), and resuspended in 60 μl of hybridisation buffer (50% formamide, 10% dextran sulphate, 0.1% Tween 20, 2×SSC and 10 mM Tris-HCl, pH 7.4) and 6 μl of yeast tRNA (100 mg/ml, Invitrogen, California, USA). Prehybridisation was carried out for 1 h at 37°C as described. To prehybridise the arrays, 80 μl of herring sperm DNA (10 mg/ml, Sigma Aldrich, Poole, Dorset, UK) and 135 μg of human Cot1 DNA (Roche) were coprecipitated and resuspended in 60 μl of hybridisation buffer. Open-well hybridisation was carried out as described previously.40 To optimise the number of arrays, the following procedure was used: in the first experiment, the array was hybridised with a mixture of patient A DNA labelled with Cy3 and patient B DNA labelled with Cy5.
Image and data analysis
After hybridisation, slides were scanned on a GenePix 4000B scanner (Molecular Devices, Sunnyvale, California, USA). Fluorescent intensities were extracted after subtraction of the local background using SPOT software.41 The data were normalised by dividing the mean ratio of each clone duplicated by the median ratio of all autosomal clones. Clones were excluded in cases where the duplicate values differed from each other by >10%. Mean and standard deviation (SD) values of hybridisation ratios for all clones in each experiment were calculated, and experiments where SD>0.1 were excluded from analysis (but were successfully repeated subsequently). We investigated those clones where the hybridisation ratio exceeded a value of the mean plus or minus four times the SD for the particular hybridisation experiment.
Fluorescence in situ hybridisation
Fluorescence in situ hybridisation (FISH) experiments were carried out to confirm deletions and duplications identified by microarray analysis, using standard techniques.42 Metaphase spreads were prepared from patient-derived lymphocytes using standard procedures. The human genomic clones from the RPCI BAC and PAC libraries were obtained from BACPAC resources (http://bacpac.chori.org).
Genotyping
Genotyping was carried out as previously described.43 Polymerase chain reaction (PCR) amplification of genomic DNA from parents and patients (100 ng) was undertaken separately. PCR products were then pooled according to their size and labelling, analysed on a capillary electrophoresis system (ABI 3130 Applied Biosystems, California, USA), and alleles were identified using GeneScan and Genotyper software.
RESULTS
Array-CGH analyses
Our series consisted of 29 patients, 17 males and 12 females, presenting with idiopathic ASD syndrome. The mean percentage of clones analysed for the whole series was 93%.
Thirty three chromosome gains or losses were detected in 22 patients. The size of the aneusomic segment varied from 200 kb (1 clone) to 16 Mb (19 clones).
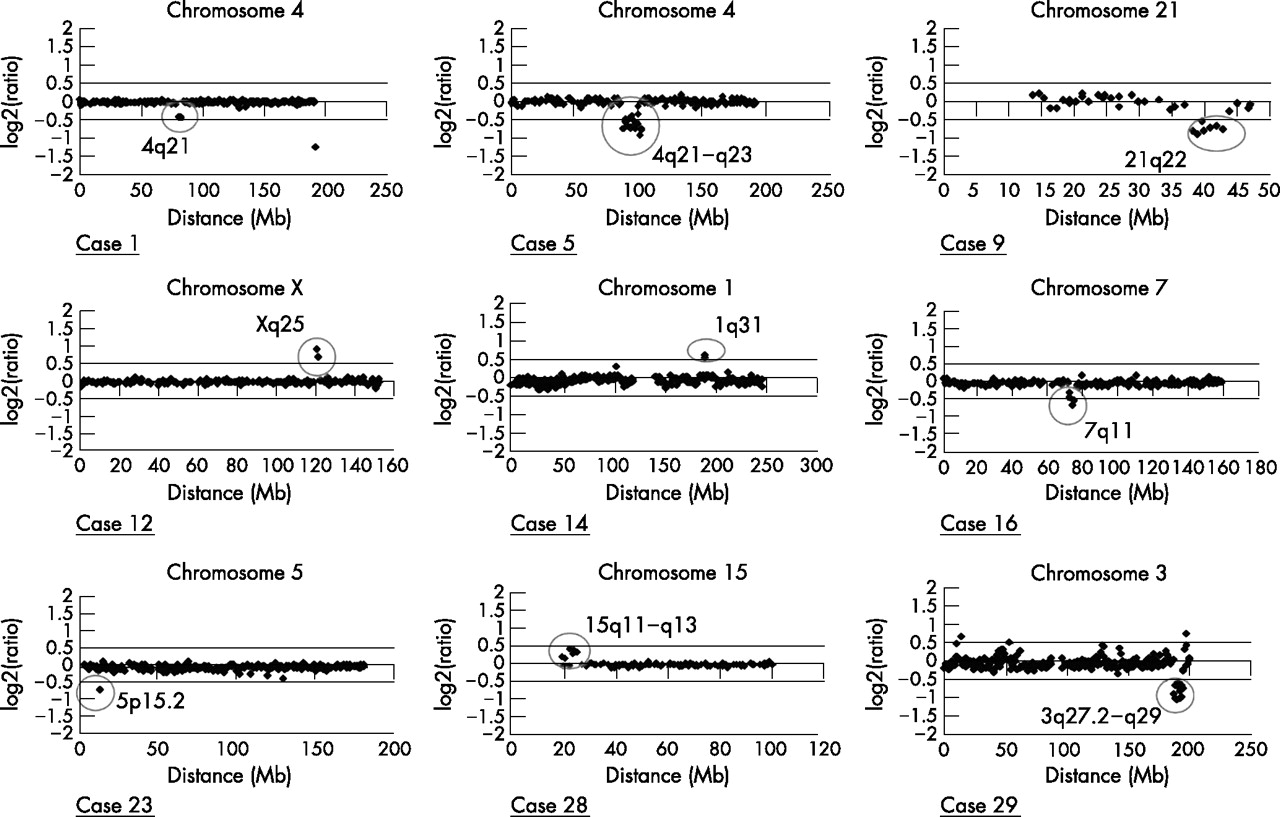
To distinguish clinically relevant anomalies from normal chromosomal variants, we first took advantage of previously published studies looking for clones with copy number variability in normal people or otherwise suspected polymorphism.44–46 Twenty three variants had previously been described as normal variants in the general population and were therefore not further analysed (table 1). The 10 remaining variants were considered to be possibly pathogenic and were further investigated (table 2). Among these, seven were deletions and three were duplications. All deletions or duplications found are in distinct non-overlapping regions. Figure 1 shows the array-CGH profiles of these cases.
Details of the polymorphisms found in the cohort of our study
Patients with possibly pathogenic chromosomal anomalies

Array-based comparative genomic hybridisation ratio profiles showing chromosomal imbalances. For each graph, the x axis marks the distance from the telomere of the short arm (Mb) and the y axis marks the hybridisation ratio represented on a log 2 scale. Horizontal lines indicate the threshold for clone deletion or duplication (mean (4 SD)). Aberrant clones are surrounded.
The 10 cases described here have been submitted to the DECIPHER database (http://www.sanger.ac.uk/PostGenomics/decipher/) providing access to detailed phenotype information and viewing of the genomic imbalances within the context of the genome browser, Ensembl (http://www.ensembl.org/index.html).
Confirmation of chromosomal anomalies by independent techniques
The 10 possibly pathogenic imbalances were then validated by at least one independent method: FISH or genotyping analysis of samples from the proband and both parents using either previously characterised microsatellites or new microsatellites identified from genomic sequence data in the critical regions. In one case (Xq25 duplication, patient 12), microarray analysis at tiling path resolution was carried out to confirm and refine the abnormal finding (data not shown). The deletion of case 5 was retrospectively visible on standard karyotype and was not further studied by FISH. In all cases, these analyses confirmed the chromosomal anomalies identified by array-CGH.
To further show the clinical relevance of these chromosomal aberrations, clinically normal parents of nine patients were tested by FISH or genotyping to discriminate between de novo and inherited anomalies. The causal relationship remains uncertain for case 14 because the de novo occurrence could not be tested as parental blood samples were not available. Seven rearrangements occurred de novo; the two remaining cases (case 12 and 23) were shown to be inherited from a normal parent. The first is a Xq25 microduplication in a boy (case 12), inherited from his healthy mother who presented an X inactivation bias. The unaffected brother does not carry this microduplication. The second, case 23, is a 5p15.2 microdeletion, ranging from 200 kb to 1.3 Mb, inherited from the healthy mother and is hence likely to be non-pathogenic.
Genotyping studies showed that 55.5% of the anomalies are of maternal origin and 45.5% paternal (table 2).
Clinical data
Table 3 summarises the clinical features of the patients (fig 2) with microdeletion or microduplication. These patients are briefly described below.
Clinical features observed in patients with clinically relevant chromosomal rearrangements

{kind=link}
{kind=link}
Facial appearance of patients with chromosomal imbalance detected by array-based comparative genomic hybridisation. (Photographs are reproduced with their parents’ consent.)
Case 1 (46,XY,del(4)(q21.1q21.22)(4.8 Mb)) is a 20-year-old man, born at term with normal neonatal measurements and Apgar score. Pregnancy was marked by a toxoplasmic seroconversion between months 5 and 8 of gestation. He presented with delayed motor milestones, congenital malformations (Meckel diverticulum, ectopic testes, hypospadias, lumbar scoliosis and abnormal tooth position) and dysmorphic facial features (convergent strabismus, short philtrum and retrognatism). He developed postnatal short stature (−4 SD). He fulfils the DSM IV criteria for autism. Cerebral computed tomography scan showed periventricular calcifications. At age 14, cerebral magnetic resonance imaging (MRI) showed cerebellar atrophy.
Case 5 (46,XY,del(4)(q21.23q24)(16.5 Mb)) is a 12-year-old boy born at term with normal neonatal measurements. He presented with axial hypotonia, arthrogryposis, ectopic microtestes, congenital hip dislocation and large atrium septum defect. Motor development was delayed. He developed scoliosis. He has a marked hypotonic face, two hair whorls, bilateral ptosis, microstomia and a broad chest. His abilities were normal in the verbal domain (VIQ 71), but less preserved in performance (PIQ 54). He fulfils the DSM IV criteria for autism. Cerebral MRI is normal.
Cases 7 and 9 (46,XX,del(1)(p36.33p36.32)(2.1 Mb)) and (46,XX,del(21)(q22.13;q22.3)(6.6 Mb)), respectively, have been reported previously.35 They fulfil the DSM IV criteria for PDD-NOS.
Case 12 (46,XY,dup (X)(q25)(3.2 Mb)) is a 24-year-old man born at term with standard neonatal measurements. At the age of 6 years, he developed a scoliosis that was surgically treated at 15 years of age. He fulfils the DSM IV criteria for autism.
Case 14 (46,XX, dup(1)(q31.1q31.2)(2.4 Mb)) is a 20-year-old woman born at term with normal neonatal measurements and Apgar score. She presented with motor milestones delay and has never been able to say a single word. Physical examination showed thin synophris and low-set ears. She fulfils the DSM IV criteria for autism. Cerebral computed tomography scan is normal.
Case 16 (46,XY,del(7)(q11.22q11.23)(4.9 Mb)) is a 24-year-old man born at term with standard neonatal measurements. The diagnosis of Williams’s syndrome had been proposed earlier in infancy but FISH study for 7 q11.2 deletion with a probe corresponding to elastin gene was normal. His motor and language milestones were delayed. He progressively developed spasticity. His growth parameters were normal. He had a hypospadias, a dorsolumbar scoliosis, and dysmorphic features with anteverted nares, long philtrum, malar hypoplasia and narrow palate. He loves music. He fulfils the DSM IV criteria for autism.
Case 28 (46,XX,dup (15)(q11.2q13.1)(4.6 Mb)) is a 25-year-old woman born at term with standard neonatal measurements and congenital hip dislocation. Her developmental milestones were delayed. She has developed epilepsy with polymorphic episodes since the age of 18 years. Her growth parameters were normal. She had dysmorphic features with a long thin face, anti-mongoloid palpebral fissures, gothic palate and dentomaxillar dysharmonia. She fulfils the DSM IV criteria for autism. Her cerebral magnetic resonance imaging was normal.
Case 29 (46,XY,del(3)(q27.2q29)(8.8 Mb)) is a 14-year-old boy born at term with standard neonatal measurements. He had a narrow pelvis and chest with pectus carinatum. He developed severe dorsal kyphosis. Since birth, he has had feeding difficulties and his weight progressively decreased to −3 SD. He presented with dysmorphic features: triangular face, deep set eyes, gothic palate, micrognathia, abnormal helix and low neck hairline. He fulfils the DSM IV criteria for PDD-NOS. Cerebral magnetic resonance imaging showed thick corpus callosum, megacisterna magna and accentuated Virchow–Robin spaces.
DISCUSSION
We report here the first study of patients presenting with syndromic ASD using whole-genome array-CGH at a 1 Mb resolution. In all, 29 patients were investigated. We identified 33 copy number variations in 22 patients; 8 were considered to be clinically relevant on the basis of
-
their association to autism in a previous report;
-
their size;
-
their de novo occurrence; and
-
their genes content.
We therefore detected a pathogenic chromosome imbalance in 27.5% of the patients who were investigated. Hence, array-CGH should be considered to be an essential aspect of the genetic analysis of patients with syndromic ASD.
Among the eight rearrangements, 1 (3.4%) is subtelomeric, whereas 7 (24.1%) are interstitial and two (ie, one quarter) are duplications. Altered segments varied in size from 1.4 to 16 Mb (2–19 clones). A single identified aberration was estimated to be >10 Mb in size, but still was not previously detected by high-resolution karyotyping, whereas seven copy number abnormalities are inferior to 5 Mb.
No recurrent abnormality was found in this cohort. These results underlie the extreme genetic heterogeneity of syndromic ASD, an observation consistent with the previously described clinical heterogeneity of these conditions. These data strongly support the idea that only a whole-genome high-resolution analysis such as array-CGH is able to provide a high diagnostic yield for chromosomal imbalance in patients with ASD.
Interestingly, we identified two chromosomal imbalances which show the phenotypic variability of chromosomal rearrangements. Indeed, patient 28, who carries the 15q11q13 duplication, had patent dysmorphic features, whereas previous publications underlie the moderate or absent facial dysmorphism in patients with 15q11q13 duplication. FISH and genotyping analyses of this anomaly show that this phenotypic difference is due to neither breakpoints position nor parental origin, and further support the hypothesis that all patients presenting with ASD should be tested for 15q11q13 anomalies. Similarly, case 16 carries a complex deletion covering the Williams’s locus. On the basis of facial dysmorphism, diagnosis of Williams’s syndrome had been evoked for this patient in infancy, but FISH analyses (Vysis and Aquarius probes) were normal. Further investigations are under way to characterise this complex rearrangement which shows that rearrangements occurring in hotspots of recombination may sometimes have unusual breakpoints and might be missed using metaphase FISH with standard probes.
Regarding possible genotype–phenotype correlations, several conclusions can be drawn from our data. Firstly, patients with duplications seem to be mildly affected compared with patients with deletion. Secondly, the severity of the mental retardation does not seem to be directly related to the size of the chromosomal imbalance, as the largest rearrangement was detected in the less severely retarded patients (case 5). Thirdly, all patients with a chromosomal imbalance are dysmorphic. The association to the ASD of a facial dysmorphism therefore seems to be a good indication for chromosomal anomaly screening.
CONCLUSION
Our data emphasise the benefits of whole-genome array-CGH at 1 Mb resolution for diagnosis of syndromic ASD, as chromosomal imbalances were detected three times more often than previously described. Further studies in larger samples of patients with ASD are needed to confirm the high incidence of cryptic chromosomal rearrangements in these patients compared with patients with syndromic mental retardation. Besides the importance of these results for diagnosis and genetic counselling, they may also pave the way for the identification of new ASD genes. Indeed, array-CGH is a powerful disease gene identification strategy, as shown by the recent identification of CHD7 gene causing CHARGE syndrome by array-CGH screening of patients diagnosed with this syndrome.47 Some of the chromosomal imbalances identified in this study may therefore point out some interesting candidate genes as—for instance, the GRIA3 (glutamate receptor subunit 3) gene contained in the duplicated region carried by patient 12. Finally, array-CGH testing of all patients before inclusion will also benefit linkage studies conducted on multiplex families to identify autism susceptibility loci. The yield of such linkage studies will increase if the cohort comprises only cases with truly idiopathic autism.
Acknowledgments
We thank the patients and their families who participated in this study.
REFERENCES
Footnotes
-
Published Online First 13 July 2006
-
Funding: This study was supported by the Centre National de la Recherche Scientifique, the Fondation de France and the Wellcome Trust. M-LJ was supported by Fondation pour la Recherche Médicale and RR was supported by a Sanger Institute Postdoctoral Fellowship.
-
Competing interests: None declared.